따라서 Random walk model 의 시간에 따른 평균은 t*mu 이고, 분산은 t*sigma^2 이다. 만약 Z의 평균이 0이라고 가정하더라도 분산이 시간에 따라 점점 커진다는 것을 알 수 있다. 따라서 Random walk model 은 stationarity 를 만족하지 않는다.
moving average 의 parameter q 와 가중치 theta 를 고정해놓고 계산을 하면, 평균과 분산은 t 와는 관계 없이 고정된다는 것을 알 수 있다. 따라서 moving average model 은 stationarity 를 만족한다.
추가적으로 Moving average model 의 auto covariance function 을 구해보자.
moving average model 은 stationarity 를 만족하기 때문에 auto covariance function 은 time spacing 에만 의존한다. 또한 이전 포스팅에서 time spacing 이 최대 q 인 경우에만 자기상관성이 존재한다는 것을 correlogram 을 통해 확인할 수 있었다. moving average model 의 노이즈의 평균이 0일 때를 가정하고 covaraicne 를 구해보자.
특정 시점 t에서의 주가를 X_t 라고하자. 또한 특정 시점 t 에서의 회사의 공지 Z_t (noise) 가 주가에 영향을 미친다고 하자. 그런데 과거 시점 (t-1, t-2...) 에 회사의 공지도 주가에 영향을 미친다. 이런 경우에 X_t 를 다음과 같이 모델링할 수 있다.
# noise 생성
noise <-rnorm(10000)
ma_2 = NULL
# ma(2) 생성을 위한 loop
for (i in 3:10000) {
ma_2[i] = noise[i] + 0.7*noise[i-1]+0.2*noise[i-2]
}
# shift
moving_average_process <- ma_2[3:10000]
moving_average_process <- ts(moving_average_process)
par(mfrow=c(2,1))
plot(moving_average_process, main = "A moving average process of order 2", ylab = "")
acf(moving_average_process, main = "Correlogram of ma (2)", ylab = "")
correlogram 을 보면 time step 이 0,1,2 인 경우에만 상관성이 있는 것을 확인할 수 있다. 우선, time step 이 0 인 경우는 항상 auto correlation coef 1이다. 또한 현재값에는 최대 2 time step 전의 noise 까지 반영이 되기 때문에, 최대 2 time step 의 값과 상관성이 있다는 것을 확인할 수 있따.
아래와 같이 정의되는 X_t 를 random walk 이라고 한다. X_t 는 이전 time step 에서의 값 X_t-1 에 noise Z가 더해진 값이다. random sampling 과 다른점은 현재값이 이전값에 더해진다는것이다. 이는 랜덤하게 어떤 한 방향으로 걷는것과 비슷하다. 매번 시작점에서 한발짝 걷는 것이 아니라 한발짝 걸어서 도착한 곳에서 다시 한발짝을 간다.
$$ X_t = X_{t-1} + Z_t $$
$$ Z_t \sim Normal(\mu, \sigma) $$
이러한 random walk 모델에서 X_t 는 이전 time step 에서의 값 X_t-1 과 매우 큰 연관성을 갖는다. 따라서 non-stationary time series 데이터이다.
Random walk model simulation in R
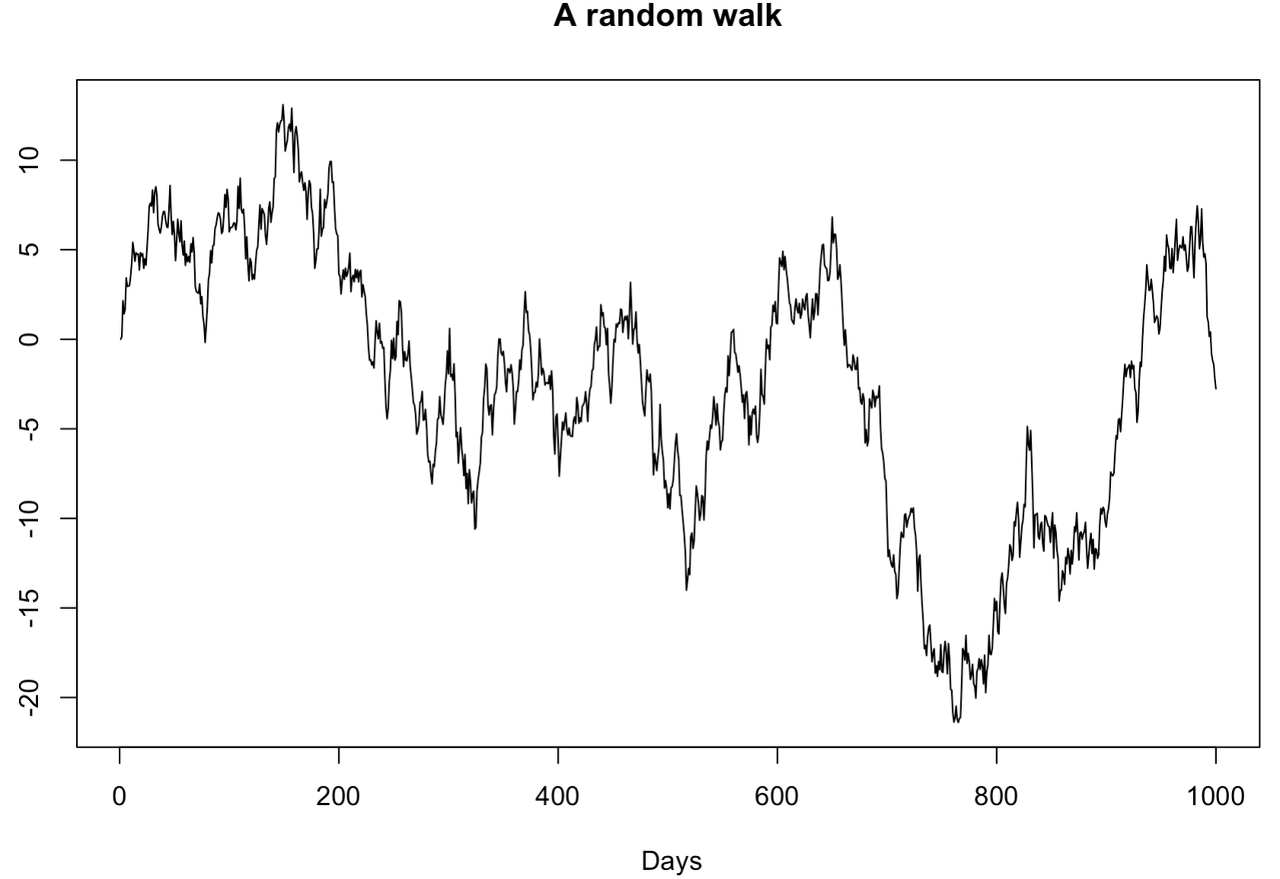
R 로 random walk 모델을 만들어보자. 아래는 1000개의 random walk 데이터를 생성하는 예제이다. 시계열 그래프를 그려보면, 이 데이터는 non-stationary time series데이터라는 것을 확인할 수 있다. 구간을 나눠서보면 트렌드를 보이기 때문이다.
x <- NULL
x[1] <- 0
for(i in 2:1000){
x[i] <- x[i-1]+rnorm(1)
}
random_walk <- ts(x)
plot(random_walk, main="A random walk", ylab="", xlab=" Days", col="black")
위 그림은 전형적인 random walk 그래프이다.
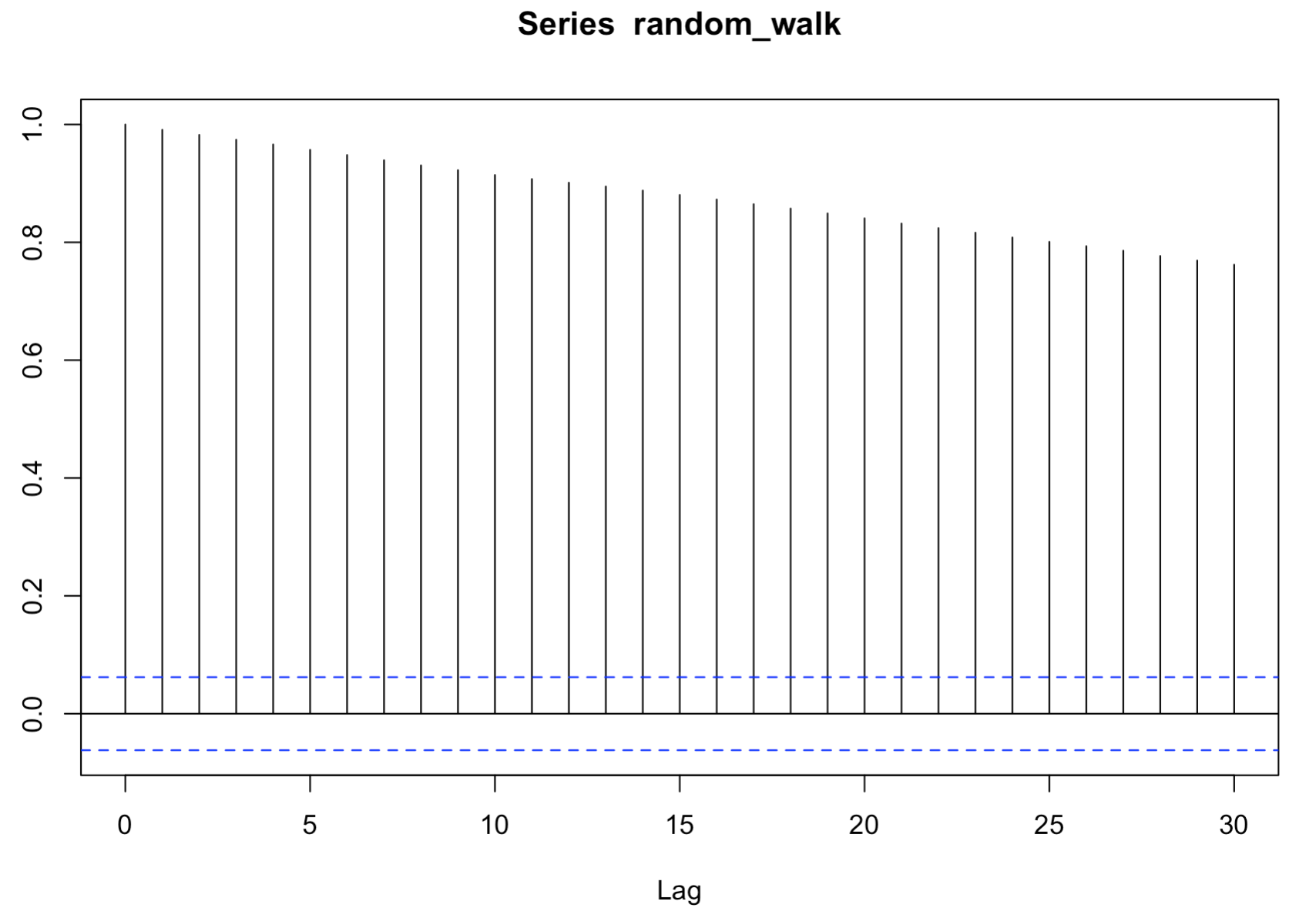
random walk 데이터에서 correlogram 을 그려보자. 인접한 time step 에서 auto correlation coefficient 가 큰 패턴을 보이기 때문에 non-stationary time series 라는 것을 다시 확인할 수 있다.
acf_result <- acf(random_walk)
random walk 모델에서 noise Z는 stationary time series 라고 볼 수 있다.
$$ Z_t \sim Normal(\mu, \sigma) $$
noise 가 stationary time series 라는 것을 데이터로 실제로 확인해보자.
random_walk_diff <- diff(random_walk)
plot(random_walk_diff, main="A random walk diff", ylab="", xlab=" Days", col="black")
앞선 포스팅에서 auto covariance coefficient 에 대해 설명하였다. auto covariance coefficient 은 time series 데이터에서의 각각의 time point 간 연관성을 의미하는데, stationary time series 에서는 k 라고하는 parameter 에 의해 달라진다. auto covariance coefficient 의 추정값 c_k 는 아래와 같이 계산된다.
이번에는 auto correlation coefficient 에 대해 정리해보려고 한다. auto correlation coefficient 도 auto covariance coefficient 와 마찬가지로 time series 데이터에서 time step 별 값의 연관성을 의미하는데 범위를 -1~1로 조정한 것으로 이해할 수 있다. 마치 공분산과 상관계수의 관계와 같다.
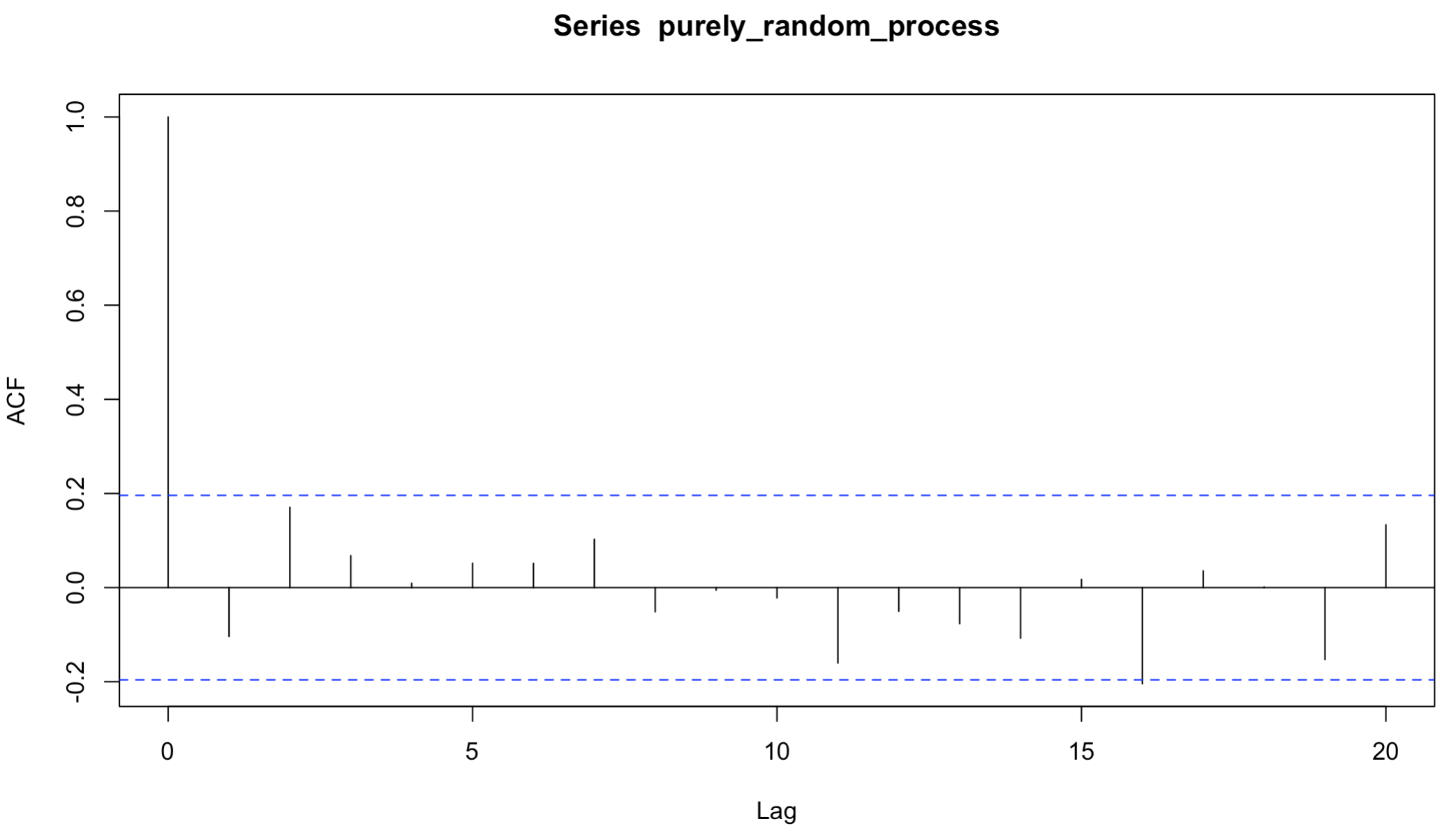
아래 R 코드는 100개의 표준정규분포를 따르는 데이터를 만든 후, correlogram 을 그리는 코드이다. 파란선은 연관성이 유의한지에 대한 임계치를 의미한다. 유의한 데이터 포인트가 하나 밖에 없고, lag 에 따른 패턴이 보이지 않으므로, 전체적으로 시계열 데이터가 자기상관성이 없다고 결론 내릴 수 있다.
실제 데이터를 correlogram 을 그려보자. 다음은 모 어플리케이션의 월간 활성 이용자수 (MAU, monthly active user) 추이이다. 이 서비스는 점점 성장하는 추이를 보여주고 있다. 시계열 데이터의 관점에서는 시간에 따른 평균의 변화 (trend) 를 보이는 non-stationary time series 이다.
위 데이터에서 correlogram 을 그리면 아래와 같이 나타난다. lag 에 따른 auto correlation coef 의 패턴이 보이며 (점점 감소), 인접한 데이터 포인트에서는 유의한 상관성을 보이고 있는 것을 확인할 수 있다.
어떤 종류의 데이터이든 상관 없으며, 그저 시간에 따라 수집된 데이터를 시계열 데이터 (timeseries data) 라고 한다.
한국의 일별 코로나19 신규 확진자수 추이
예를 들어, 일별 코로나 확진자수는 1일이라고 하는 time step 으로 수집된 시계열 데이터의 한 종류이다.
Week stationary time series
week stationary time series 란 다음의 조건을 만족한다.
1) 시간에 따른 평균 (mean) 에 변화가 없다.
2) 시간에 따른 분산 (variance) 의 변화가 없다.
3) 주기적인 등락 (flucation) 이 없다.
이러한 조건을 만족하긴 위해서는 time series 의 한 섹션 (A 섹션) 고른 후, 다른 섹션 (B 섹션) 을 골랐을 때, A, B 섹션이 비슷하면 된다.
Stochastic process
random variable 의 collection - X1,X2,X3 .. 가 있다고 하자. 이들이 각각 다른 모수를 가진 분포를 따를 때, 이를 stochastic process 라고 한다. stochastic process 의 반대개념은 deterministic process 이다. deterministic process 는 모든 step (t) 에 대해서 예측 가능하다. 예를 들어, 어떤 함수에 대한 미분함수는 특정 X 에서의 Y 값을 정확하게 알 수 있다. 이와 반대로 stochastic process 는 매 step 이 random 이기 때문에 어떤 확률 분포에서 왔다는 것만을 알 수 있을 뿐, 값을 정확하게 예측할 수 없다.
$$ X_t \sim distribution(\mu_t, \sigma_t) $$
예를 들어, 다음과 같은 시계열 데이터가 있다고 해보자.
$$ X_1 = 30, X_2 = 29, X_3 = 57 ... $$
시계열 데이터를 바라보는 한 가지 관점은 stochastic process 의 실현 (realization) 으로 보는것이다. 매 timestep 별로 어떤 확률 변수가 정해지고 우리는 그 확률변수에서 나온 하나의 샘플값을 관찰하는 것이다.
Autocovariance function
stationary time series 라는 가정을 하자. 두 가지 timestep s,t 에서의 covariance 를 정의해볼 수 있다. (확률 변수이기 때문)
$$ \gamma(s,t) = Cov(X_s, X_t) $$
$$ \gamma(s,s) = Var(X_s) $$
또한 아래처럼 covariance function 을 정의할 수 있는데, 이 함수는 stationary time series 라는 가정 하에 t 에 따라서는 값이 바뀌지 않으며, k가 결정하는 함수가 된다. (아래 식에서 c 는 추정값이다.) 이러한 time step (k) 에 따른 공분산의 식을 autocovariance function 이라고 한다.
$$ \gamma_k = \gamma(t, t+k) \sim c_k $$
즉, stationary time series 에서는 Cov(X1, X2) 나 Cov(X10,X11) 이나 기댓값은 같다고 할 수 있다. 그 이유는 데이터에서 두 가지 섹션을 선택했을 때, 그 모습이 똑같다고 기대하는것이 stationary time series 이기 때문이다.
또한 gamma(t_k, t) 는 autocovariance coefficient 라고 하며, stochastic process 에서의 실제 autocovariance 값이다. 데이터를 통해 구한 c_k 를 통해 autocovariance coefficient 를 추정한다.
Autocovariance coefficient
그러면 Autocovariance coefficient 의 추정값은 어떻게 구할까? timestep 을 k 라고 할 때, 추정값은 아래와 같다.
Apache Pig 는 data processing 을 위한 플랫폼으로 Pig Latin 이라는 high-level language 를 통해 HDFS 데이터에 접근한다. 이 때, execution environment 로 Map Reduce, Tez 등을 가질 수 있다. 또한 in built operation (minimum, average 와 같은) 을 다양하게 지원한다. 이러한 다양한 function 을 지원하면, 여러가지를 Pig 환경에서 실행할 수 있기 때문에 더욱 효율적이라고 할 수 있다. Pig 의 활용분야는 다음과 같다.
cloudera VM 에서 다음과 같은 명령어를 입력하면 pig 의 interactive shell 인 grunt 로 접속하게 된다. 위 커맨드는 /etc/passwd 폴더를 hdfs 에 저장하고, map reduce 를 execution engine (back-end) 으로 해서 pig 를 실행한다는 뜻이다.
grunt > A = load '/user/cloudera/passwd' using PigStorage(':');
grunt > B = foreach A generate $0, $4, $5 ;
grunt > dump B;
load 명령어를 통해 해당 file 을 hdfs 에서 load gkrh, foreach 에서 subset 을 extract 한다 (각 line 마다, 1,5,6번째 컬럼을 가져옴).
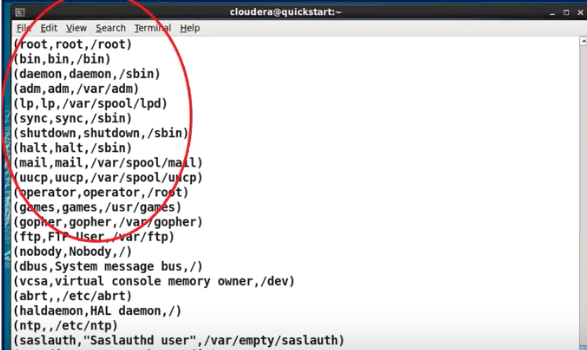
결과는 /user/cloudera/passwd 에 저장된 정보의 일부를 위 그림처럼 출력하게된다. 결과를 파일형태로 저장하기 위해서는 아래와 같은 명령어를 수행할 수 있다.
grunt > store B into 'userinfo.out'
2) Apache Hive
Apache Hive 는 HDFS 에 대한 SQL interface 인 HiveQL 을 통해 데이터를 manage, query 하는 플랫폼이다. 이러한 과정을 beeline 이라는 command line interface 를 통해 interactive 하게 수행할 수 있다. Hive 는 기본적으로 Data warehouse software 이다. execution environment 로는 Map reduce, Tez, Spark 를 가질 수 있다. HDFS, HBase 에 저장된 데이터를 다룰 수 있고, mapper, reduce 를 customization 할 수 있다.
앞선 pig 예제와 마찬가지로 /etc/passwd 를 hdfs 에 저장하고 beeline 커맨드를 실행한다.
CREATE Table userinfo (uname STRING, paswd STRING, uid INT, gid INT, fullname STRING, hdir STRING, shell STRING) ROW FORMAT DELIMITED FIELDS TERMINATED BY ':' STORED AS TEXTFILE;
LOAD DATA INPATH '/tmp/passwd' OVERWRITE INTO TABLE userinfo;
SELECT uname, fullname, hdir FROM userinfo ORDER BY uname;
위 커맨드는 userinfo 라는 table 을 생성하고, 파일로부터 데이터를 읽어들여 select 문을 통해 출력하는 예제이다.
본 예제에서는 beeline command 를 사용했지만 이외에도 Hive command line interface, Hcatalog, WebHcat 도 있다.
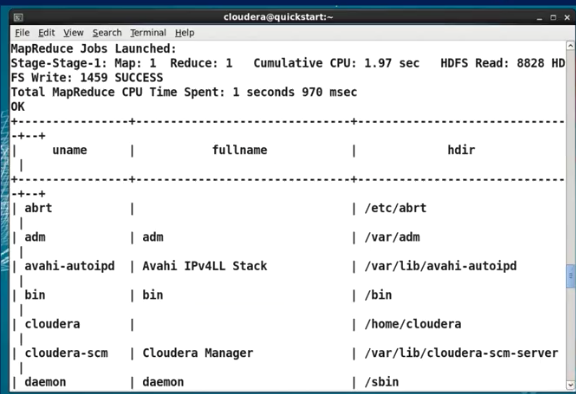
3) Apache HBASE
Hbase 는 scalable data store 이며 non-relational distributed databased 이다. HDFS 위에서 동작하며 In-memory operation 을 제공한다. 이는 cache 와 같은 개념이기 때문에 더 빠르게 같은 일을 처리할 수 있다. Hbase 의 특징은 1) replication 2) security 3) SQL like access 이다.
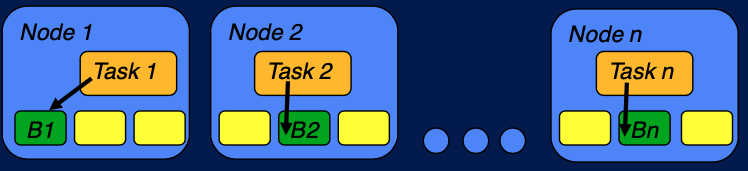
이전 포스팅에서 Map reduce 프레임워크의 한계에 대해 설명하고, 이를 보완하기 위하여 YARN, Tez, Spark 등이 사용된다는 것에 대해 간단하게 언급하였다. 하둡 아키텍쳐에 대해 다시 remind 를 해보자.
Hadoop architecture
위 그림과 같이 데이터가 node 에 분산되어 저장되어있고, task 가 data node 에 할당되어 처리를 한 후에, 이 결과를 merge 하는 것이 하둡이 어떤 job 을 실행하는 방법이다. 이것이 이전 포스팅에서 언급한'계산을 데이터로 보내는 것' (computation to data)의 개념이라고 볼 수 있다. 이러한 방법은 불가능한 것을 가능할 수 있고, 무엇보다 더욱 효율적으로 (빠르게) job 을 처리할 수 있다. 병렬적으로 수행할 수 있고, 데이터의 이동이 최소화되기 때문이다. 하지만 문제는, MapReduce execution framwork 는 Map reduce paradigm 을 구현할 수 있는 application 에만 유용하다. 어떤 application 이 map reduce 를 통해 구현할 수 없는 경우에 문제가 생긴다. 이를 보완하기 위하여 YARN, Tez, Spark 등이 사용된다.
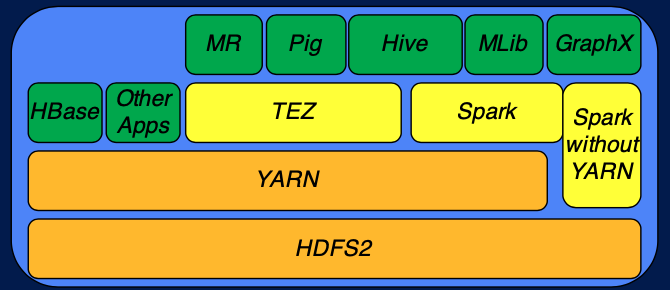
Next Generation Execution Framworks
YARN 은 기본적인 Hadoop 의 execution engine 이다. 이 위에 YARN 위에서 동작하는 HBase 와 같은 application 이 있다. 또한 YARN 위에서는 Tez 와 Spark 와 같은 다른 framework가 올라갈 수도 있다. Pig 나 Hive 같은 application 은 TEZ 와 Spark (Hive 의 경우 TEZ와 Spark 모두 이용) 위에서 동작한다. 또한 Spark 는 YARN 없이도 동작할 수 있는 execution framework 이다.
Tez 의 특징은 dataflow graph 를 지원하고, custom data type 을 지원한다. 따라서 맵 리듀스 프레임워크에서 처럼 모든 데이터의 입출력이 key-value pair 로 이루어져야하는 제약이 없다. Tez 를 활용하는 것의 장점은 자원을 효율적으로 관리하고 복잡한 태스크를 DAG (directed acyclic graph) 를 활용해서 실행할 수 있다는 것이다.
Hive on Tez example
Hive 는 backend execution engine 으로 Tez 를 이용하는 것을 지원한다. 예를 들어 아래 코드를 보자.
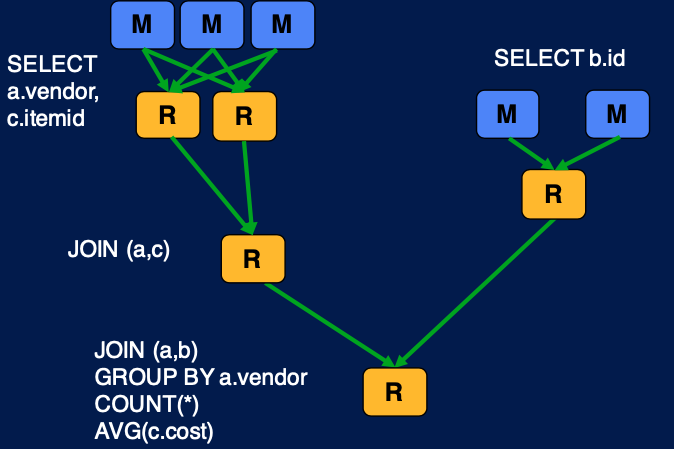
SELECT a.vendor, COUNT(*), AVG(c.cost) FROM a
JOIN b ON (a.id = b.id)
JOIN c ON (a.itemid = c.itemid)
GROUP BY a.vendor
1) Original Hadoop MapReduce 를 사용한 해법
위의 쿼리는 여러개의 map reduce job 으로 나뉘어진다. 하나의 map reduce job 이 다른 job 의 인풋이 되고, 이것들이 조합이 되어 최종적인 쿼리의 결과를 가져올 수 있다.
2) Tez 를 사용한 해법
Tez 를 사용하면 조금 더 간단하게 같은 작업을 수행할 수 있다. original map reduce 만을 사용했을 때와의 차이점은 intermediate map task 가 없다는 것이다. map reduce 의 경우 원래는 결과를 hdfs 에 저장하도록 되어있는데, 그러지 않고 데이터를 재사용함으로써 graph 를 간편화할 수 있고 이를 통해 효율적으로 같은 job을 수행할 수 있는 것이다.
Spark 의 경우 advanced DAG execution engine 이며 cyclic data flow 를 지원한다. 또한 in-memory computing 을 지원한다. 우선 데이터 처리를 memory 위에서 할 수 있기 때문에 매우 빠르고, DAG 사이에 데이터 공유도 가능하다는 장점이 있다. 또한 Java, Scala, Python, R 언어를 지원하기 때문에 범용성이 높은 execution engine 이라고할 수 있다. 예를 들어 Spark python interface 를 통해 Logistic regression 을 수행하는 코드를 확인해보자.
points = spark.textFile(...).map(parsePoint).cache()
w = numpy.random.ranf(size = D) # current separating plane
for i in range(ITERATIONS):
gradient = points.map(
lambda p: (1 / (1 + exp(-p.y*(w.dot(p.x)))) - 1) * p.y * p.x
).reduce(lambda a, b: a + b)
w -= gradient
print "Final separating plane: %s" % w
original map reduce 는 iterative data process 에 적용하기 매우 힘들다는 단점이 있었다. 바로 위 코드에서 iteration 이 등장하는데, 같은 데이터셋을 이용해서 gradient 를 여러번 반복해서 구해서 weight 를 업데이트하는 logistic regression 의 training 과정이다. 우선 같은 데이터셋을 여러번 반복해서 사용하기 때문에 .cache() 함수를 통해 데이터를 RAM 에 저장시킨다. 위 코드는 spark 공식 홈페이지의 예제 (http://spark.apache.org/examples.html)인데 original map reduce 와 100배 정도의 속도차이가 난다고 한다. 이러한 이유로 in-memory computing 이 가능한 경우 선호된다. 이 링크에서는 다양한 Spark 예제를 확인할 수 있다. 또한 spakr 는 Machine learning 을 위한 라이브러리도 제공하기 때문에 이를 직접 구현하지 않고도, high-level interface 를 사용할 수도 있다 (https://spark.apache.org/docs/2.1.0/ml-classification-regression.html).
References
코세라 - Hadoop Platform and Application Framework 강의를 참고하였습니다.
docker-compose up -d
docker-compose exec hive-server bash
docker-compose up -d 는 각각의 docker-compose.yml 에 위치한 container 를 background 실행하는 명령어
The docker-compose up command aggregates the output of each container (essentially running docker-compose logs -f). When the command exits, all containers are stopped. Running docker-compose up -d starts the containers in the background and leaves them running.
CREATE TABLE employee
(
name string,
work_place ARRAY<string>,
sex_age STRUCT<sex:string,age:int>,
skills_score MAP<string,int>,
depart_title MAP<STRING,ARRAY<STRING>>
)
ROW FORMAT DELIMITED
FIELDS TERMINATED BY '|'
COLLECTION ITEMS TERMINATED BY ','
MAP KEYS TERMINATED BY ':';
--Verify tables creations run in Beeline
!table employee
--Load data
LOAD DATA LOCAL INPATH '/HiveBook/Chapter_03/employee.txt' OVERWRITE INTO TABLE employee;
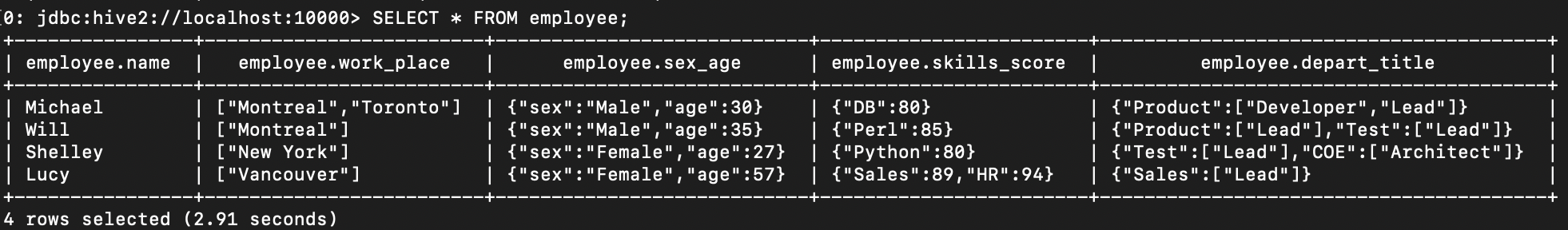
--Query the whole table
SELECT * FROM employee;