전체 글 (321)
-
2024.05.08
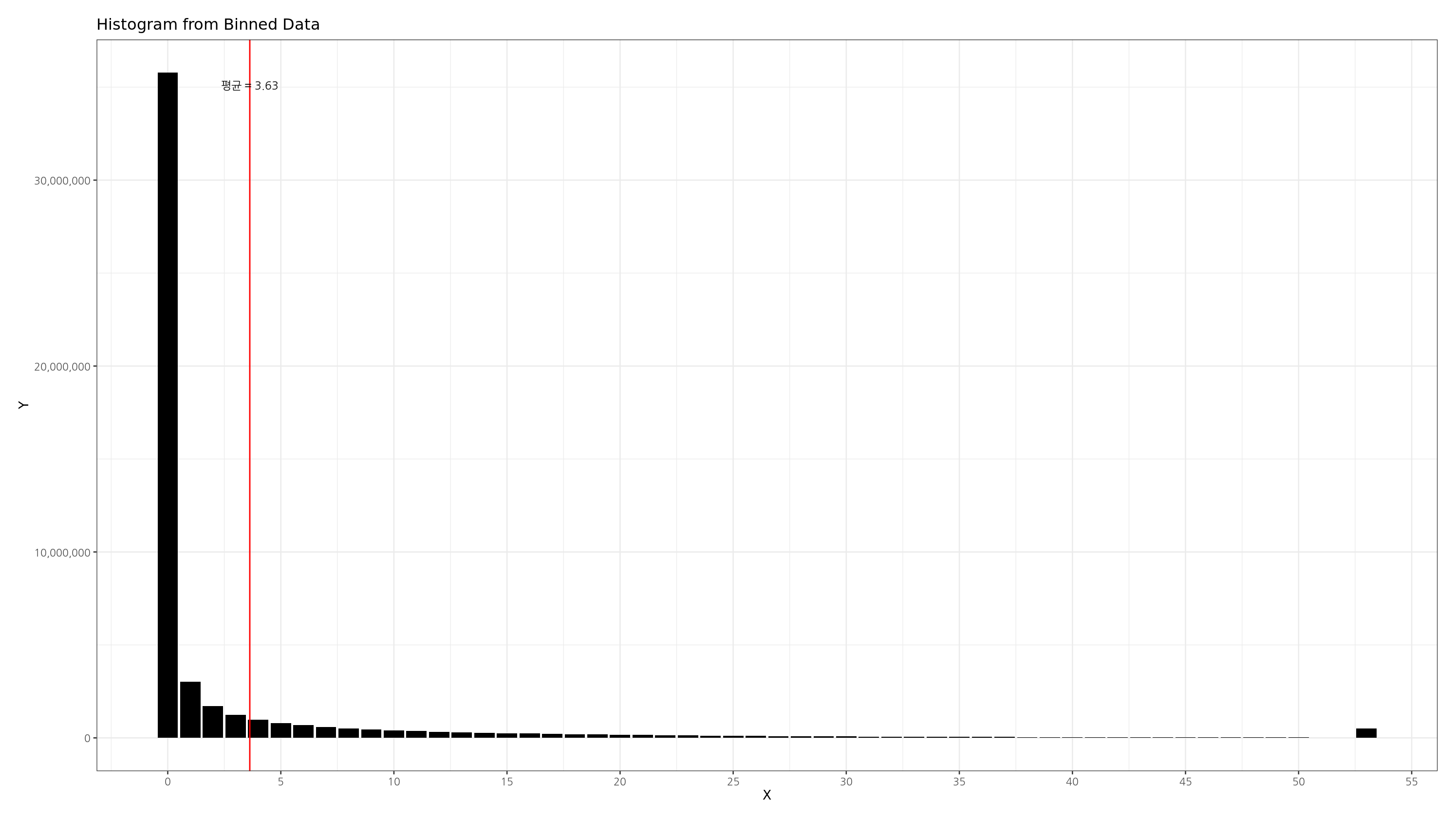
R - 롱테일 분포의 히스토그램 그리기
-
2024.05.07
일반화 선형 모형의 개념 및 회귀 계수의 의미
-
2024.05.07
선택 편항과 Collider bias 에 대한 설명
-
2024.05.07
코크란-멘텔-헨젤 검정과 공통 오즈비의 추정 방법
-
2024.04.27
유전학에서의 딥러닝 활용이 정밀의학에 어떻게 기여하는가? -
2024.04.26
rstudio server 에서 github copilot 사용하기 -
2024.04.04
R 에서 폰트 사용하는 방법 (linux) -
2024.03.30
범주형 분석 - 교차표에서 효과를 추정하는 방법 -
2024.03.06
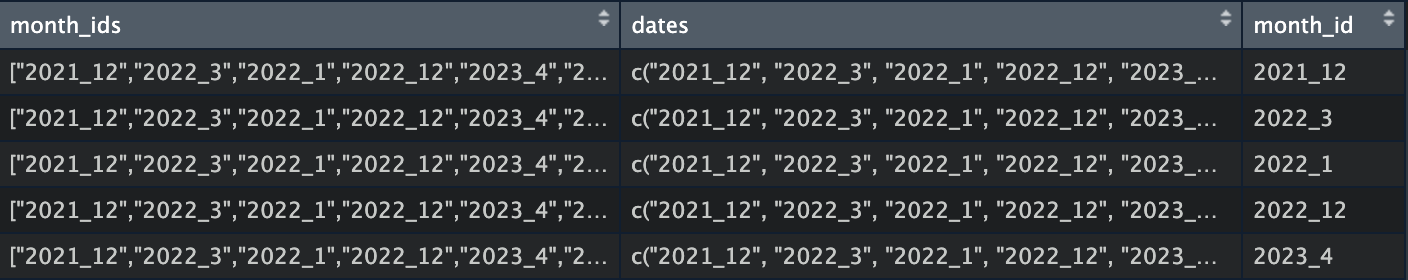
R - 리스트 문자열을 벡터로 바꾸고 unnest 하기 -
2024.03.05
omitted variable bias 의 방향을 알 수 있는 팁