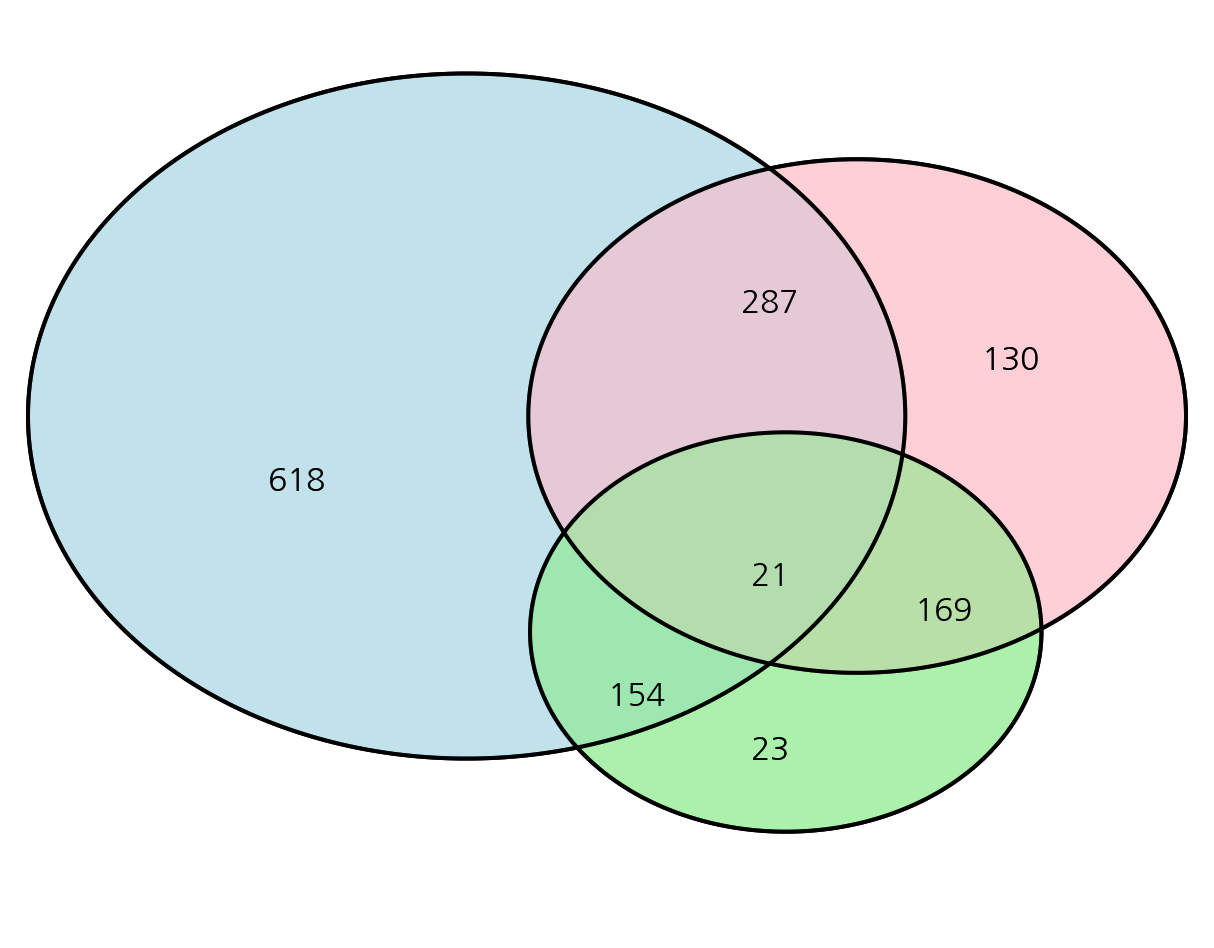
어떠한 두 그룹의 교차되는 부분의 규모를 표현할 때, 벤다이어그램은 매우 효과적인 방법입니다. 벤다이어그램을 그리기 위해 파워포인트, Keynote 같은 프로그램을 이용할 수 있습니다. 하지만, 실제 수치를 반영해서 원의 크기를 조정하고 싶을 수 있습니다. 이 때, R 의 VennDiagram 패키지를 이용할 수 있습니다.
코드는 아래와 같습니다. draw.pairwise.venn 함수에 area1, area2, cross.area 에 해당하는 값을 넘겨줍니다. 이 때 주의할 점은 area1, area2 에서는 교차되는 부분을 포함하는 값을 넣어주어야 합니다.
VennDiagram 패키지를 통한 벤다이어 그램 그리기
2개 집합 벤다이어그램
area1 : A
area2 : B
cross.area : A∩B
library(VennDiagram)A<- 1000B<- 400C<- 100
# 벤다이어그램 원 내부 수치 있도록 변경 # scaled = FALSE 로 지정하면, 실제 수치를 고려하지 않은 벤다이어그램을 그려줌grid.newpage()v<- draw.pairwise.venn(area1 = A, area2 = B, cross.area = C, fill = c("light blue", "pink"), alpha = rep(0.5, 2),fontfamily='Kakao Regular', scaled=TRUE)grid.draw(v)
# cex = 0 옵션을 통해 벤다이어그램 원 내부 수치 제거grid.newpage()v<- draw.pairwise.venn(area1 = A, area2 = B, cross.area = C, fill = c("light blue", "pink"), alpha = rep(0.5, 2),fontfamily='Kakao Regular', cex=0, scaled=TRUE)grid.draw(v)
1. Koch's Postulates: 특정 미생물이 특정 질병의 원인임을 증명하기 위한 4가지 기준
질병이 발생한 모든 사례에서 미생물이 발견되어야 한다.
해당 미생물을 순수 배양해야 한다. 특정 질병의 원인 미생물이 어떤 것인지 정확히 확인하기 위해서는 다른 미생물이 섞이지 않은 상태에서 해당 미생물만을 배양해야 한다.
배양한 미생물을 건강한 숙주에 접종하여 동일한 질병을 유발해야 한다.
실험적으로 감염된 숙주에서 동일한 미생물을 재분리해야 한다.
질병인 경우 미생물 발견. 순수 배양 한뒤에, 건강한 사람에게 질병 유발하는지 확인. 질병 감염된 사람한테서 그 미생물이 또 발견되는지 확인. 이러면 미생물이 질병의 원인이라고 볼 수 있다.
2. Contagion(컨테이전)은 전염성 질병이 사람 사이에 전파되는 현상을 의미한다. 이 용어는 질병의 확산과 전파 과정을 설명하는 데 사용된다.
3. 만약, 환경으로부터 감염되는 질병이라고하면 이는 non-contagious 이다. 예를 들어, 라지오넬라병이나, 곰팡이(fungi) 에 의한 감염은 환경으로부터 감염되는 것이기 때문에 contagious 가 아니다.
4. Infectious Respiratory Aerosol (감염성 호흡기 에어로졸) 은 호흡기 질환을 일으킬 수 있는 병원체(바이러스, 박테리아 등)를 포함한 미세한 액체 또는 고체 입자를 의미한다.
- Droplet transmission : 비말 전파 : 액체로 부터 직접적으로 접촉해서 감염되는 경우. 비말이 빠르게 지면으로 침강하기 때문에 이로 인한 전달은 빠른 시간내에 이루어짐. 비교적 큰 입자 크기 (5마이크로미터 이상)
- Airborne transmission (droplet nuclei transmission) : 비말핵 전파 : 집적 접촉이 아니라, 비말핵이 공기중을 떠돌아다니다가 감염. 감염자와 직접적으로 같이 있지 않더라도, 감염자가 머문 공간의 공기에 접촉해서 감염될 수 있음. 비교적 작은 입자 크기 (5마이크로미터 이하)
5. Obligate Airborne Transmission(필수 공기 전파) 정의: 병원체가 주로 공기를 통해서만 전파되는 경우를 말한다. 이러한 병원체는 공기 중에 떠 있는 미세한 에어로졸 입자를 통해 전파되며, 이는 감염된 사람과 직접 접촉하지 않고도 쉽게 전파될 수 있다 (pulmonary TB).
6. Preferentially Airborne Transmission(선호 공기 전파) 정의: 병원체가 여러 전파 경로 중에서 공기 전파를 선호하지만, 반드시 공기 전파만으로 전파되지는 않는 경우를 말한다. 이러한 병원체는 공기 전파 외에도 비말 접촉, 표면 접촉 등을 통해 전파될 수 있다 (chicken pox).
7. Airborne viral infections 의 예시 (공기 전파 바이러스 감염)
- 일반 감기
- 인플루엔자
- epidemics 에 의한 분류: pandemic vs. seasonal
- host 에 따른 분류 : non-zoonotic vs. zoonotic - 주의사항: 일부 동물병의 경우 드물게 사람을 감염시킬 수 있다.
- 코로나바이러스
8. Airborne bacterial infections 예시 (공기 전파 박테리아 감염)
- Mycobacterium tuberculosis 균에 의한 Pulmonary tuberculosis (TB, 결핵) 감염
- Legionella 균에 의한 Legionallosis, Pontiac fever 발생
- TB 는 사람간 전파됨 (contagious)
- 레지오넬라는 사람간 전파안됨 (non-contagious)
- Anthrax (탄저병) : 탄저균(Bacillus anthracis) 감염에 의해 발생하는 급성 감염질환. 감염된 동물에 의해 사람에게 감염됨 (non-contagious)
9.Fungal infection (mycosis) : 곰팡이나 효모 같은 진균류에 의해 발생하는 감염
- pathogenic fungi 를 포함하고 있는 공기를 흡입하여 발생함
10. Allergy : 면역 반응에 의한 Hypersensitivity reaction 이다. 알러지를 일으키는 물질을 Allergen 이라고함.
- Allergic asthma : Cough and wheezing
- Allergic rhinitis : Running nose
- Allergic bronchopulmonary mycoses : Fungal allergy, infection in lungs
... 등등 Allergy 에는 다양한 분류와 증상이 존재한다. 감염과 allergy 는 다르다. 곰팡이는 감염을 일으킬 수도 있고, 알러지를 일으킬 수도 있고, 둘 다 일으킬 수도 있다.
11. Thunderstorm asthma : Thunderstorm Asthma(천둥번개 천식) 는 천둥번개를 동반한 폭풍우가 발생한 후 특정 기상 조건에서 많은 사람들이 급성 천식 발작을 일으키는 현상을 말한다. 이 현상은 주로 봄과 여름철에 발생하며, 특히 호주와 같은 일부 지역에서 잘 알려져 있다.
원인: 폭풍우가 발생하기 전에 공기 중에 높은 농도의 꽃가루와 곰팡이 포자 (plant pollen, fungal spores) 가 존재할 수 있다. 이들 알레르겐은 폭풍우의 강한 바람과 번개에 의해 작은 입자로 분해되며, 비와 함께 대기 중에 분산된다.
12. 다른 알러지 발생 요인
- House dust mite (집먼지 진드기) 알러지 : 99% 가 HDM 의 feces 에 의해 발생
- Cockroaches : saliva, feces, shedding skin 등에 의해 발생
- Dog : major source - saliva, dander, urine
- Cat : saliva, fur, urine
- Bird : excrements, feather dust (깃털에 존재하는 1마이크로미터 이하의 매우 작은 물질이 있음)
13. Bioactive compounds 에 의한 건강 영향
- endotoxins : gran-negative bacteria 에서 나오는 독성
- mycotoxins : fungi 로 부터 나오는 독성 (대사 산물임)
생명체로부터 나오는 독성물질을 의미함
14. 어린 시절 개와 같이 사는 등 endotoxins 과 같은 알러지, 질병 유발 요인에 노출되는 것이 면역체계 발달에 좋은 영향을 줄 것이라는 가설이 존재함
15. HEPA filter (High Efficiency Particulate Air Filter) : 필터가 가장 포집하기 어려운 입자가 대략 0.3마이크로미터 입자인데, 이는 입자가 크거나 작으면 각기 다른 포집 방법에 의해 잘 포집되는데, 애매한 크기는 각기 다른 포집방법에서 잘 포집되지 않기 때문이다. HEPA 필터는 0.3 마이크로미터 입자의 99.7% 이상을 포집할 수 있어야한다.
16. 공기청정기의 성능: CADR (Clean Air Delivery Rate)
V = volume of the test chamber (ft3)
k_e = decay rate mearsured with the air purifier (min-1)
k_n = natural decay rate measured without the air purifier (min-1)
CADR=V(ke−kn)
16. HVAC System : Heating, Ventilation, and Air Conditioning (HVAC) system : 온도 조절, 실내 공기질을 좋게 유지시켜주는 시스템
17. UVGI (UltraViolet Germicidal Irradiation) : UVGI는 자외선의 살균 특성을 이용하여 공기, 물, 그리고 표면의 미생물을 불활성화시키는 방법
- In-duct UVGI : HVAC system 안에 설치해서 흐르는 공기를 자외선을 이용해 살균함
- Upper room UVGI: 방 상단에 설치해서 실내 공기를 살균함
- 자외선중에 UV-C 라는 자외선이 사용된다.
18. Greenhouse gases (GHGs) : 대기의 열을 잡아놓는 기능을 하는 가스
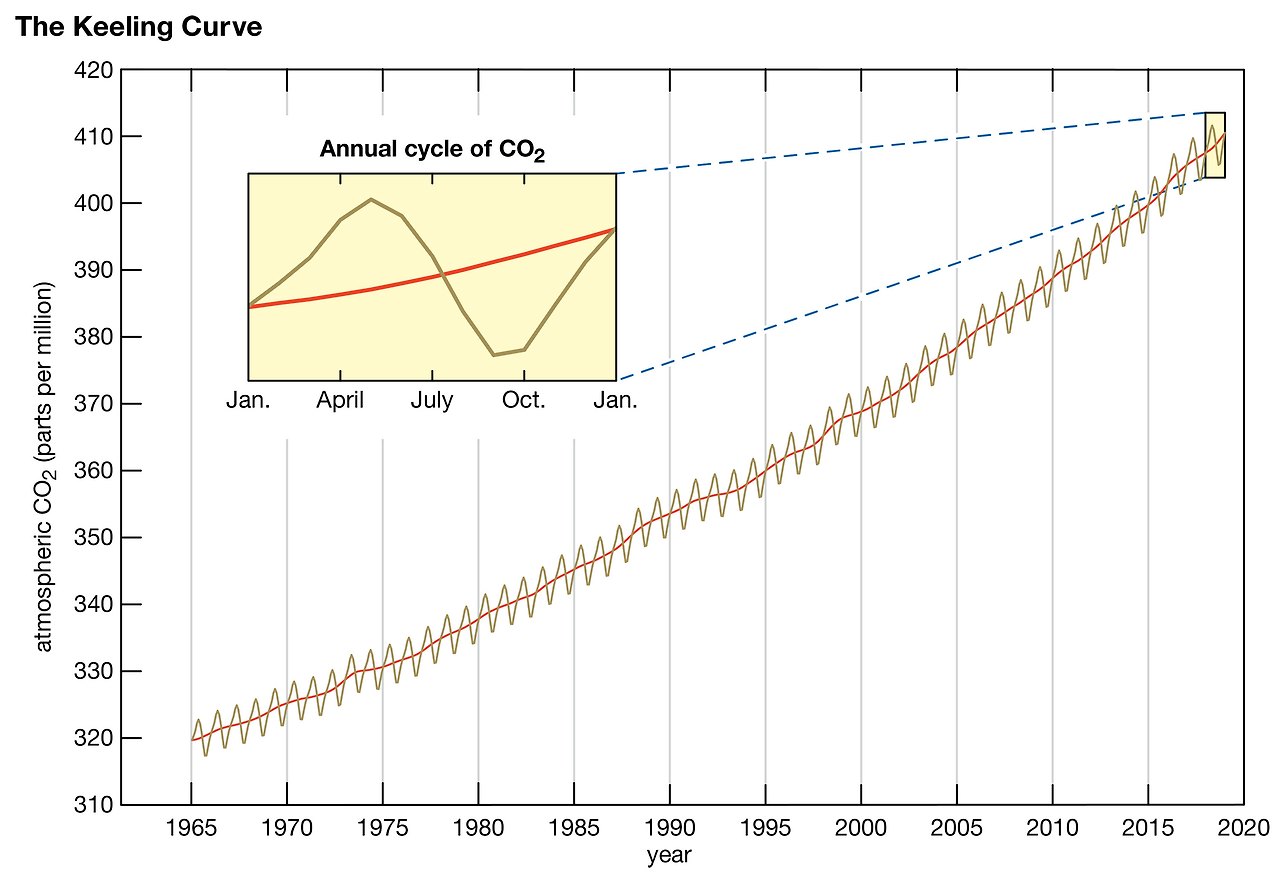
19. The Keeling Curve : 킬링 곡선은 지구 대기의 이산화탄소(CO₂) 농도를 시간에 따라 나타낸 그래프다. 이 곡선은 1958년에 하와이의 마우나로아 관측소에서 대기 중 CO₂를 체계적으로 측정하기 시작한 찰스 데이비드 킬링(Charles David Keeling)의 이름을 따서 명명되었다. 이 그래프는 인간 활동이 지구 기후에 미치는 영향을 보여주는 중요한 지표 중 하나다.
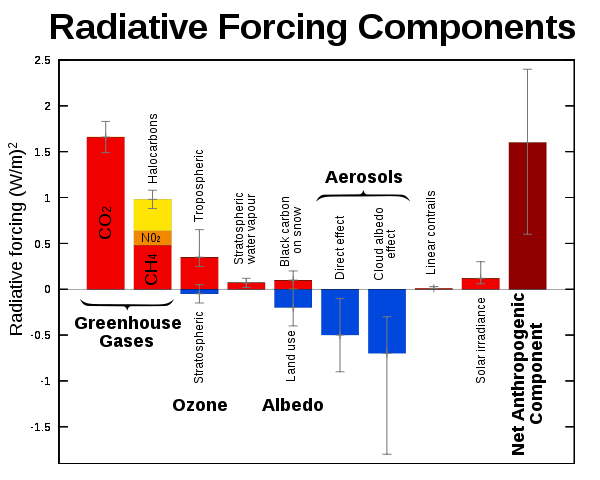
20.Radiative forcing : 복사 강제력(radiative forcing)은 지구의 에너지 균형에 영향을 미치는 요인을 측정하는 개념이다. 이는 지구 대기로 들어오는 태양 복사 에너지와 지구에서 방출되는 적외선 복사 에너지 간의 균형 변화를 나타낸다. 복사 강제력은 기후 변화를 이해하고 예측하는 데 중요한 역할을 한다.
위 그림에서 Radiative force 가 +인 요인은 지구 온도를 상승시키는 요인. -인 요인은 지구 온도를 하락시키는 요인임
21. Aerosol-cloud-radiation interactions
에어로졸은 태양 복사 및 지구 복사를 직접적으로 흡수하거나 반사하여 지구의 복사 균형에 영향을 미친다:
직접 효과 (Direct Effect):
에어로졸 입자는 태양 복사를 반사하여 지표면으로 도달하는 태양 에너지를 감소시킨다. 예를 들어, 황산염 에어로졸은 태양 복사를 반사하여 냉각 효과를 나타낸다.
일부 에어로졸은 태양 복사를 흡수하여 대기를 가열한다. 예를 들어, 검댕(black carbon)은 태양 복사를 흡수하여 온난화 효과를 나타낸다.
간접 효과 (Indirect Effect):
에어로졸은 구름의 반사율과 수명을 변화시켜 지구 복사 균형에 간접적으로 영향을 미친다. 예를 들어, 에어로졸이 많은 구름은 반사율이 높아져 냉각 효과를 나타낼 수 있다.
하지만 실제로 지구 온도에 대한 aerosol 의 영향을 정확하게 측정하기 어렵다. 많은 부분이 불확실하게 남아 있음.
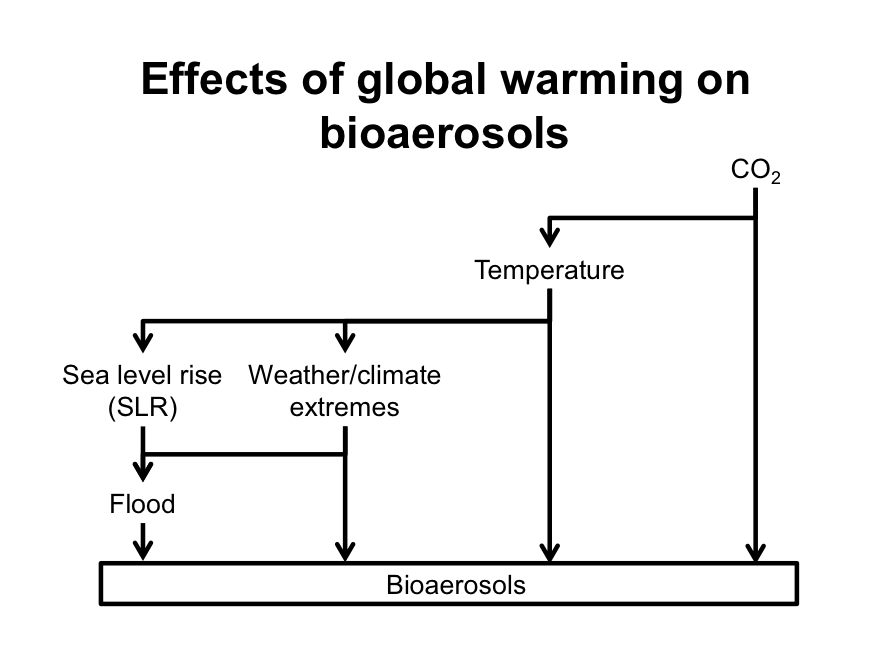
22. 지구 온난화가 bioaerosol 에 미치는 영향 : Global warming 인 아래와 같은 경로로 bioaerosol 에 영향을 미친다.
- 이산화탄소 상승 -> 기온 상승 -> 식물 성장 증가, 꽃가루 증가
- 이상 기후 증가 -> 천둥 천식
- 해수면 상승 -> 범람 증가 -> 쓰나미가 일어난 이후 대기 오염으로 인해 천식이 증가하는 사례 (Katrina cough)
23. Bioaerosol 이 세계 기후에 미치는 영향 : 구름 형성, 복사 강제력 (열의 흡수나 반사를 통해 지구 온도를 변화시키는 힘)
- 구름형성: 에어로졸은 대기에서 cloud condensation nuclei (CCN) 또는 ice nuclei (IN) 처럼 작용할 수 있음
- 구름 응결핵(CCN)은 수증기가 응결하여 구름 방울을 형성하도록 도와주는 입자다. 에어로졸에 수증기가 붙어 액체가 되면서 성장하는 방식으로, 수증기상태의 물이 액체 상태로 변경하도록 도와줌.
- 얼음핵(IN)은 대기 중의 수증기가 얼음 결정으로 변하는 과정을 촉진하는 입자다.
24. Bioprecipitation : 대기 중 특정 미생물, 특히 박테리아가 구름 형성과 강수(비, 눈) 과정에서 중요한 역할을 한다. 그리고 비에 섞여 있다.
1. PCR 발명자는 폴란드 생화학자 Kary Mullis 이다. (PCR = polymerase chain reaction : DNA 의 특정 부분을 증폭시키는 기술)
2. 찰스 다윈은 비글호에서 케이프 베르데 제도 근처에서 먼지를 수집했는데, 이를 사하라 먼지라고 한다. 이는 먼지가 매우 넓은 지역에 걸쳐 이동할 수 있다는 것을 의미하고, 먼지가 대기 중에서 얼마나 멀리 퍼질 수 있는지를 보여주는 중요한 사례이다.
3. 코흐는 1880년대에 감자 조각과 이후에는 한천을 이용한 고체 배지 (solid culture media) 를 개발하여 세균을 배양하는 방법을 개선했다. 이는 세균학 연구에서 중요한 진전을 이뤄냈으며, 다양한 세균을 분리하고 연구하는 데 큰 기여를 했다.
4. 한천 배지(Agar Media)와 젤라틴 배지(Gelatin Media)는 미생물 배양에 사용되는 두 가지 주요 고체 배지이다. 둘 중, 한천 배지가 일반적으로 더 많이 사용된다.
5. LIF : Laser Induced Fluorescence(레이저 유도 형광) : 레이저를 이용해 입자나 분자에서 형광을 유도하여 공기 중의 생물학적 입자나 물질을 검출하고 분석하는 기술
6. MALDI는 Matrix-Assisted Laser Desorption/Ionization의 약자이다. 이는 질량 분석법에서 사용되는 이온화 기술 중 하나로, 주로 단백질, 펩타이드, 당, 폴리머 등의 큰 분자들을 분석하는 데 사용된다. 최종적으로 질량 대 전하 비율(m/z) 을 통해 어떤 입자인지를 특정할 수 있다.
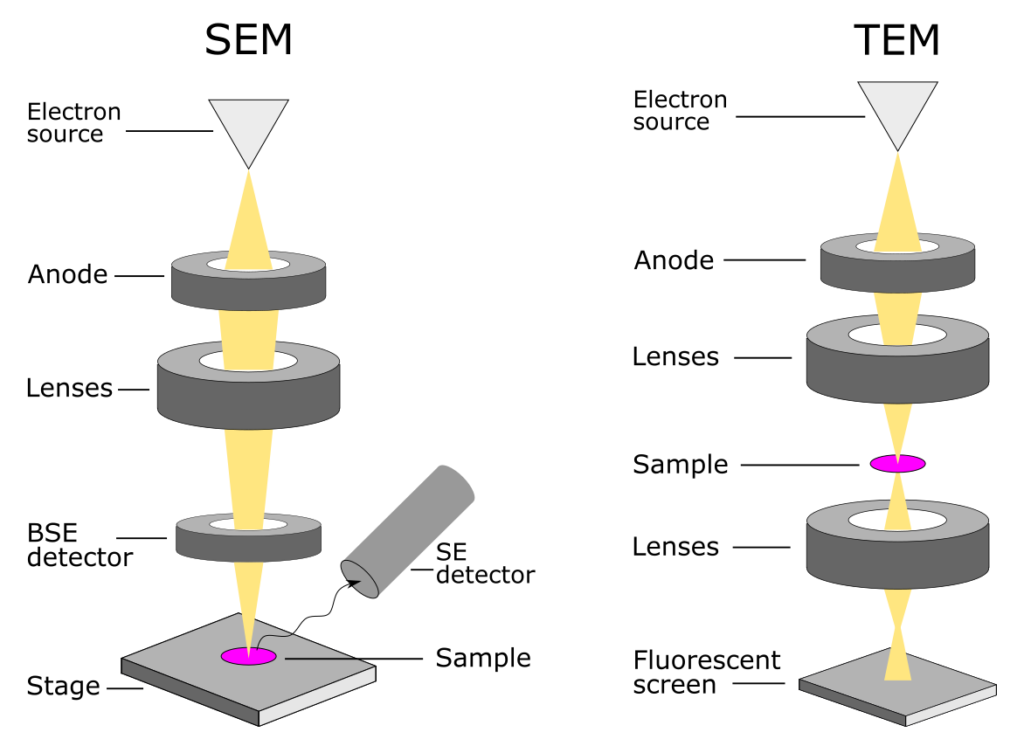
7. TEM은 Transmission Electron Microscopy(투과 전자 현미경)의 약자이다. TEM은 전자 현미경의 한 종류로, 고해상도의 이미지와 세부 구조를 관찰하는 데 사용된다.
8. 인플루엔자 A 형: H1N1 ~ H18N11 까지 다양한 형으로 존재함 (신종플루, 스페인독감)
인플루엔자 B 형: 두 가지 주요 라인(B/Victoria와 B/Yamagata)으로 나뉨
9. Legionella (레지오넬라)는 주로 물 환경에서 서식하는 박테리아로, 레지오넬라 폐렴(Legionnaires' disease)과 폰티악 열(Pontiac fever)을 일으킬 수 있다. 레지오넬라 감염은 일반적으로 사람 간 전파(person-to-person transmission)가 아닌 환경적 노출을 통해 발생한다.
10. 생명체의 분류 (domain of life) : 생명체는 Eukaryote(진핵생물)와 Prokaryote(원핵생물)로 구분되고, Prokaryote 는 Archaea(고세균)와 Bacteria(진정세균) 으로 분류됨
11. terminal settling velocity : 종단속도 = 공기저항에 의한 힘과 중력이 평형을 이루어 속도가 일정하게 하강하는 상태를 의미함.
relaxiation time 은 종단속도에 이르기까지 걸리는 시간을 반영하는 지표인데 (실제로 걸리는 시간은 아니다.) 종단속도를 중력가속도 g로 나누어 계산함
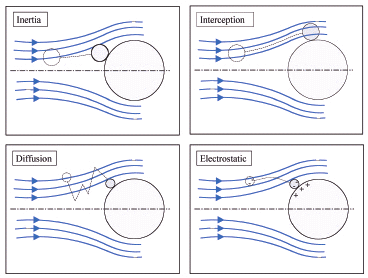
12. 필터의 3가지 원리 : Inertia, Interception, diffusion (Brownian motion)
13. 공기저항은 하강중인 입장의 직경, 모양, 속도에 의존적이다.
14. real-time bioaerosol monitor 에서 모니터링하는 biofluorophores 의 대표적인 예는 NADH 이다.
역할: NADH는 생물체의 세포 내에서 중요한 역할을 하는 보조 인자로, 주로 세포 호흡과 에너지 대사 과정에서 전자 운반체로 작용
형광 특성: NADH는 자외선(UV) 빛을 흡수하고 청색 형광을 방출하는 특성을 가지고 있다.
16. Mycobacterium tuberculosis 의 generatio time 은 1200분이다 .만약, 하나의 세포가 3일 동안 배양되었을 때, cell 의 개수는?
N=N02n
3일 = 60 * 24 * 3 = 4320m
1∗24320/1200=12.12
17. ELISA(Enzyme-Linked Immunosorbent Assay)는 특정 단백질, 항원, 항체 등을 검출하고 정량화하는 데 사용. 효소가 결합된 항체를 이용해 검출하는 방법
18. endotoxin 을 검출하기 위한 LAL (리무스 아메보사이트 리세이트, Limulus Amebocyte Lysate) 분석법은 엔도톡신을 감지하는 데 널리 사용되는 민감하고 정확한 생물학적 검사 방법
19. endotoxin 은 gram-negative bacteria 가 죽거나 용해될 때 발생하는 물질로, 강력한 면역 반응을 유발하며, 고열, 염증, 쇼크, 심한 경우 사망에 이를 수 있음
20. Air sampling (single-stage viable impactor, 400nozzles) 를 통해 airborne bacteria 샘플링을 했다. air flow rate = 28.3L/minute 이고, 3분동안 수집했다. 일정시간 배양 이후, 250 colonies 가 관찰되었다. sampled airvolume 과 bacterial concentration 을 CFU / m3 단위로 구하라. (CFU = colony forming unit)
sampled air volume = flow rate * time = 28.3L/m * 3m = 84.9L
250 colonies 에 대한 positive hole correction = 392CFU
positive hole correction 의 경우, 기기의 종류, 노즐수에 따라 정해진 값이다.250 콜로니가 관찰됐을 때, 실제 콜로니 수에 대한 값을 통계적으로 계산한 값이다.
concentration = corrected CFU / sampled air volume = 392 / 84.9L = 4.617 / L
그런데, 1L = 0.001m3 이다. (1L 는 0.1m * 0.1m * 0.1m 정육면체에 해당하는 부피). 따라서 1000을 곱해주면 4617CFU/m3 이다.
21. 바이러스는 enveloped 와 non-enveloped 로 나눌 수 있다. 외피가 있냐 없냐의 차이로, 바이러스의 특성 (감염 가능성, 환경 변화에 취약한지 등) 에 영향을 미친다. SARS 나 인플루엔자 바이러스는 Enveloped virus 이다.
sparkly 에서는 spark 에서 실행하기 위한 함수들이 존재한다. 물론 그룹별 집계 등의 기본적인 데이터 처리의 경우, dplyr wrapper 를 통해 추가적인 학습 없이도 바로 사용할 수 있지만, 이외의 데이터 처리의 경우 sparklyr 의 고유 함수에 대한 학습이 필요하다.
본 포스팅에서는 10분위수를 기준으로 변수를 10개의 카테고리로 분류하는 bucketing 작업을sparklyr 을 통해 해보고 기본 R 과 비교해보았다.
sparklyr 에서 bucketing 하기
get_spark_connection 의 경우 spark connection 을 잡는 함수인데, 관련해서 이 포스팅을 참고하길 바란다. 이 데이터처리에서 중요함수는 sdf_quantile 과 ft_bucketizer 이다. sdf 는 spark dataframe 의 약자이고, ft는 feature transformation 의 약자이다.
10분위수 구하기: sdf_quantile 을 사용하면 병렬적으로 변수의 quantile 을 구해서 리턴해준다. 이 작업은 데이터를 메모리로 로드후에 R 의 기본 quantile 을 실행하는 것보다 빠르다.
bucket 만들기: ft_bucketizer 는 연속형 변수에 대해 bucketizing 을 해준다. 이 때, 만약 10개의 bucket 으로 나눈다고 하면, splits 에 원소가 11개인 벡터를 전달해주어야한다. 10개의 bucket 으로 나눈다고 하면, 9개의 split point 가 필요한데, 여기에 최소값과 최대값을 추가로 더해 11개라고 볼 수 있다. quantile 함수에서 9개의 decile 과 최대값을 구했으므로, 최소값인 0을 추가로 더해 split 함수에 넣어준다.
bucket 이름 만들기: ft_bucketizer 함수에는 아쉽게도 bucket 의 label 을 지정하는 방법이 없다. 단지 index 만 만들어줄 뿐이다. 따라서 index 와 label 을 매핑하는 데이터프레임을 따로 만들어서 해결해볼 수 있다. quantile 의 개수만큼 seq_along 을 통해 인덱스를 만들고, names 함수를 통해 벡터의 이름을 구해서 bucket_df 라는 데이터프레임을 만든 후에, 이를 데이터와 조인하여 bucket 의 label 을 만들 수 있다.
tidy evaluation 의 경우 rlang 의 {{}} (curly-curly) operation 을 사용하는 방법입니다. 그런데, 이 방법은 함수 안에서만 사용할 수 있습니다. 즉, tidy evaluation 도 동일한 기능을 수행하지만, 아래와 같이 함수 안에서만 사용할 수 있다는 제한점이 있습니다.
이진 분류 모델의 최종 예측 값은 일반적으로 0~1사이로 나오게된다. 특정 임계치를 기준으로 테스트 양성과 테스트 음성을 분류한다. 예를 들어, 임계치를 0.5로 잡는다면, 0.5 이상인 경우를 테스트 양성, 0.5 미만인 경우를 테스트 음성으로 정의한다. 이 방법을 통해 아래와 같은 2x2 테이블을 만들 수 있다.
양성 (Disease)
실제 음성 (No Disease)
테스트 양성 (Positive)
50
10
테스트 음성 (Negative)
5
100
True Positive (TP): 50
False Positive (FP): 10
False Negative (FN): 5
True Negative (TN): 100
Sensitivity = Recall (민감도)
sensitivity 는 실제 질병인 사람 중에 테스트 양성인 사람의 비율이다.
-> 50/55 = 0.909
Specificity = Negative Recall (특이도)
specificity 는 실제 질병이 아닌 사람 중에 테스트 음성인 사람의 비율이다.
-> 100/110 = 0.909
Positive Predictive Value = Precision (양성 예측도, PPV)
ppv 는 양성으로 예측한 사람 중에 실제 질병인 사람의 비율이다.
-> 50/60 = 0.833
Negative Predictive Value (음성 예측도, NPV)
npv 는 음성으로 예측한 사람 중에 실제 질병이 아닌 사람의 비율이다.
-> 100/105 = 0.952
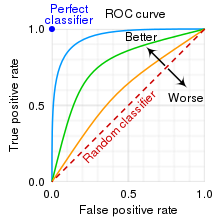
ROC 커브와 AUC (Area under curve)
임계치를 변화시키면서 1-specificity, sensitivity 그래프를 그린 것이 ROC 커브이다. 위 두 지표를 통해 그래프를 그리는 이유는 sensitivity 와 specificity 간에 트레이드오프관계가 있기 때문에, 이 관계를 시각적으로 표현하여 모델의 성능을 평가하기 위해서이다.
1-specificity = False Positive Rate (FPR)
sensitivity = True Positive Rate (TPR)
임계치가 낮아지면 모델은 더 많은 사례를 양성으로 분류하게 되어 True Positive Rate와 False Positive Rate가 모두 증가한다. 반대로, 임계치가 높아지면 모델은 더 적은 사례를 양성으로 분류하게 되어 True Positive Rate와 False Positive Rate가 모두 감소한다.
두변수가 nested 관계에 있다는 것은 full model, reduced model 관계에 있다는 것을 의미한다.
Model1:g(πi)=β0+β1x1i
Model2:g(πi)=β0+β1x1i+β2x2i
만약 모델 2에서 beta2가 0인 경우, 모델1 이 된다. 따라서 두 모델은 nested 관계에 있다.
이 경우 변수가 많은 모델2가 무조건 likelihood 가 높게 된다.
이 때, Likelihood ratio 가 카이제곱분포를 따르게된다. 자유도는 두 모델의 모수 개수의 차이가 된다. 여기서 L0가 간소한 reduced 모델이고, L1이 full model 이다. 이는 full model 과 reduce model 의 log likelihood 의 차이의 2배이다.
−2ln(L0L1)=−2(lnL0−lnL1)∼χ(1)
만약 위 통계량이 유의미한 카이제곱 값을 가지면, full model 이 reduced model 보다 좋은 것이다. 따라서 full model 을 채택한다. 만약 카이제곱값의 p-value 가 0.05보다 크다면, full model 이 reduced model 보다 좋지 않은 것이므로, reduced 모델을 채택한다.
2) 두 변수가 unnested 관계 일 때
--> AIC 가 작은 모델을 선택한다.
Model1:g(πi)=β0+β1x1i
Model3:g(πi)=β0+β1x2i+β2x3i
이러한 경우 AIC 를 통해 두 모델 중 어떤 모델이 좋은지를 판단할 수 있다. p는 변수의 숫자로 패널티텀이다. unnested 관계일 때, likelihood ratio test 를 적용할 수 없는 이유는 unnested 관계일 때는 likelihood ratio 가 카이제곱분포를 따르지 않기 때문이다.
AIC=−2(LogL1−p)
만약 모델1 의 로그 우도가 -120 이고, 모델3의 로그 우도가 -115 라고하자. 모델3의 로그 우도가 더 높다.
모델1의 AIC = -2(-120 - 2) = 244
모델3의 AIC = -2(-115 - 3) = 236
만약 모델3의 추정 파라미터 수가 2개였다면, AIC 는 234였을 것이다. 파라미터로 인한 패널티 2점이 들어갔음을 알 수 있다. 이 경우 패널티를 고려해도 모델3의 AIC가 낮기 때문에 모델3을 채택한다.
실무를 하다보면 롱테일 분포를 많이 접하게 됩니다. 예를 들어서, 어떠한 이커머스 서비스에서 "구매 금액" 이라는 변수를 살펴보면, 대부분의 유저는 구매금액이 0~1만원 사이에 들어있지만, 일부 유저는 구매금액이 몇 백만원 심지어는 몇 억원에 이르는 경우를 심심찮게 볼 수 있습니다. 극심한 right-skewed 분포 (또는 롱테일 분포)의 예라고 볼 수 있습니다.
이러한 롱테일 분포에 일반적인 히스토그램을 적용하게 되면 꼬리가 너무 길어져 가시성이 좋지 않습니다. 이런 경우에 특정 cutff 지점을 정해 따로 범주를 만들곤 합니다. 예를 들어, 구매금액이 백만원 이상인 유저는 '100만원 이상' 이라는 bucket 을 따로 만드는 것이죠. 꼬리 부분이 너무 길기 때문에 이 부분을 따로 모으는 것입니다.
R 코드로는 다음과 같이 작성해볼 수 있습니다. 포인트는 raw 데이터에 적용하는 geom_hist 를 사용하는 것이 아니라, 집계 데이터를 먼저 만든 후, geom_bar 를 통해 히스토그램을 그리는 것입니다. 그리고, 집계 데이터를 만들기 위해 cut 함수를 사용합니다.
R 코드
anal_table 데이터 프레임의 value 컬럼이 histogram 을 그리고자하는 변수입니다.
top_1_percent <- quantile(anal_table$value, 0.99, na.rm=T) # 상위 1% 경계값 찾기# bucket size 동적으로 설정
bucket_size <- 10^ceiling(log10(top_1_percent)) # 초기 bucket size# while loop를 통해 bucket size 조정while(TRUE) {
breaks <- seq(0, top_1_percent, by = bucket_size)# 상위 1% 까지의 bucket if(length(breaks) > 100) break# bucket 개수가 100개 이상이면 loop 탈출
bucket_size <- bucket_size / 10# bucket size 재조정
}
labels <- breaks
cutoff <- max(labels)+bucket_size
# 기본적으로 break 에서 좌측을 포함하지 않고 우측을 포함함(include lowest 를 통해 가장 좌측은 포함)# right=FALSE 를 통해 우측을 포함하지 않게 지정
anal_table$bucket <- cut(anal_table$value, breaks = seq(0, cutoff, by = bucket_size),
include.lowest = TRUE,
right=FALSE,
labels = labels)
# bucket 이 없는 경우는, cutoff 이상인 경우로, 따로 만든 bucket 에 속하도록 바꾸어줌
anal_table <- anal_table %>% mutate(bucket = if_else(is.na(bucket), as.character(ceiling(cutoff)), bucket))
anal_table$bucket <- factor(anal_table$bucket, levels = c(labels, ceiling(cutoff)))
summary_data <- anal_table %>% group_by(bucket) %>% count()
summary_data
summary_data <- summary_data %>% mutate(var_name = var_name)
val_quantile <- quantile((anal_table %>% select(value) %>% pull), probs=seq(0.1, 1, 0.1))
quantile_keys <- names(val_quantile)
quantile_values <- unname(val_quantile)
df_quantile <- data.frame(t(quantile_values))
colnames(df_quantile) <- quantile_keys
df_avg <- anal_table %>% summarize(avg = mean(value))
df_quantile <- cbind(df_quantile, df_avg)
df_quantile <- df_quantile %>% mutate(var_name = var_name)
total_ticks <- 10
breaks <- pretty_breaks(n = total_ticks)(range(as.numeric(as.character(summary_data$bucket))))
ggplot(summary_data, aes(x = as.numeric(as.character(bucket)), y = n)) +
scale_y_continuous(labels = scales::label_comma()) +
geom_bar(stat = "identity", fill = "black") +
labs(x = "X", y = "Y") +
scale_x_continuous(breaks = breaks, # breaks는 pretty_breaks를 사용해 계산된 값
labels = breaks) + # labels도 breaks를 사용
theme_bw(base_size = 10, base_family = "Kakao Regular") +
ggtitle("Histogram from Binned Data") +
theme(plot.margin = margin(0.5, 0.5, 0.5, 0.5, "cm")) +
geom_vline(aes(xintercept = df_quantile$avg), colour = "red") +
annotate("text", x = df_quantile$avg, y = max(summary_data$n),
label = paste("평균 =", round(df_quantile$avg, 2)),
vjust = 2, color = "black", size=3)
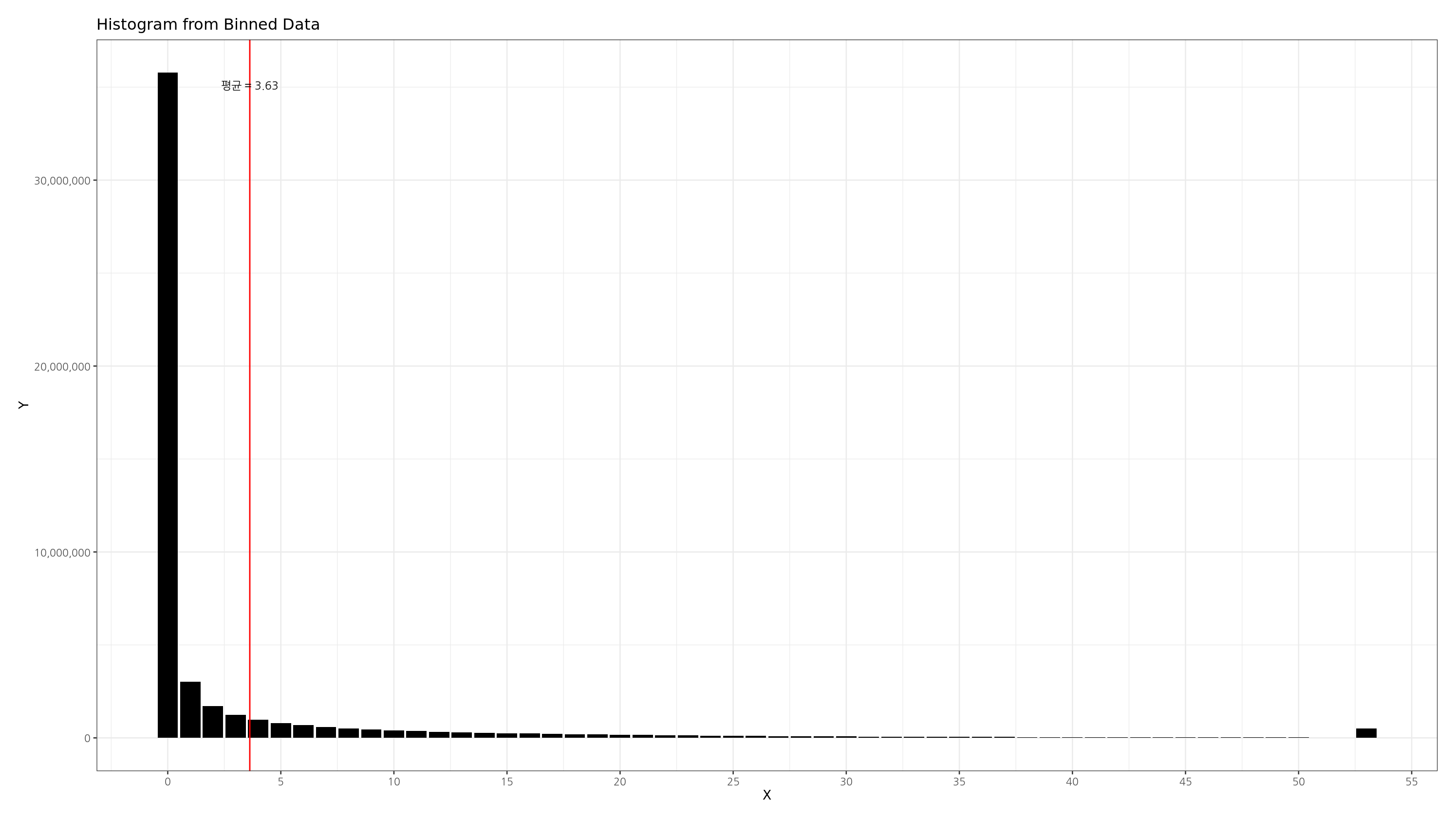
결과 히스토그램
위 코드를 통해 아래와 같이 지정된 bucket size 를 가지며, 상위 1% 이상은 하나의 bucket 으로 묶은 깔끔한 히스토그램을 그릴 수 있습니다.
위 코드에는 몇 가지 포인트가 있습니다. bucket size(bin)과 xtick 의 개수를 동적으로 결정한 부분인데요. 이 부분 코드를 좀 더 살펴보겠습니다.
bucket size 를 동적으로 결정하기
bucket 의 개수가 최소 100개가 되도록 하며, bucket size 가 1, 10, 100, 1000 처럼 10의 지수형태로 만드는 방법은 아래와 같습니다.
또한 cut 함수의 labels 를 통해 label 을 이쁘게 만들어줍니다. 예를 들어 label 이 100이라고 하면, (100~199 사이의 bucket 을 의미하는 등)
# while loop를 통해 bucket size 조정while(TRUE) {
breaks <- seq(0, top_1_percent, by = bucket_size)# 상위 1% 까지의 bucket if(length(breaks) > 100) break# bucket 개수가 100개 이상이면 loop 탈출
bucket_size <- bucket_size / 10# bucket size 재조정
}
labels <- breaks
cutoff <- max(labels)+bucket_size
# 기본적으로 break 에서 좌측을 포함하지 않고 우측을 포함함(include lowest 를 통해 가장 좌측은 포함)# right=FALSE 를 통해 우측을 포함하지 않게 지정
anal_table$bucket <- cut(anal_table$value, breaks = seq(0, cutoff, by = bucket_size),
include.lowest = TRUE,
right=FALSE,
labels = labels)
동적 xtick 의 결정
아래 코드는 총 xtick 의 개수를 10개로 고정시키고, 변수에 따라 동적으로 xtick 간격을 조정하는 코드입니다. scalse 라이브러리의 pretty_breaks 라는 함수를 사용합니다.
x1, x2 ... 가 주어졌을 때, Y를 예측하고 싶다. 근데 특정 조건하에서 Y 는 정해진 값이 아니라 어떤 분포를 따른다고 가정하고, 그 평균을 예측하고 싶을 때, 일반화 선형 모형을 활용한다. 기본적인 회귀분석에서는 반응변수가 정규분포를 따른다고 가정하고 모델링하는데, 일반화 선형 모형은 Y가 다른 분포를 따르는 경우에도 활용할 수 있는 모델링 방법이라고 볼 수 있다.
일반화 선형모형에서는 반응변수가 어떤 분포를 따른다고 가정하기 때문에 랜덤성분 (random component) 이라고 부르고, 반응변수의 평균을 설명하기 위한 설명 변수들의 함수 (위 식에서 우측부분) 를 체계적 성분 (systematic component) 이라고 부른다. 랜덤성분과 체계정 성분을 연결하는 함수를 연결함수(link function) 라고 부른다.
Y가 정규분포를 따른다면, 평균값이 -무한대~+무한대일 수 있고, Y 가 베르누이 분포를 따르면 Y의 평균이 0~1사이의 값이다. 따라서 적당한 연결함수를 통해 값의 범위를 변환하는 것이 필요하다.
또한, 일반화 선형 모형에서는 Y 가 지수족 분포를 따른다고 가정한다. 지수족 분포에는 정규분포, 이항분포, 포아송분포, 감마분포 등이 있다. Y가 따른다고 가정한 분포에 따라 알맞는 연결함수를 적용해준다. GLM 에서 지수족 분포가 중요한 개념이지만, 다소 심플하게 내용을 설명하기 위해 지수족 관련 내용은 설명하지 않겠다.
만약, Y가 정규분포를 따르는 경우에 가장 기본적으로 항등함수를 이용할 수 있다. 연결함수가 항등함수인 경우, 일반 선형 모형이라고 한다. (general liner model) (generalied linear model 과 다르다.).
μ=α+βx
연결함수가 항등함수인 경우 beta 값의 해석은 매우 쉽다. "X가 1단위 증가했을 때 반응 변수가 beta 만큼 증가한다" 고 해석한다.
Y가 베르누이분포를 따르는 경우 0~1의 값을 무한대로 변환하는 연결함수로 여러가지를 이용할 수 있다. 가장 대표적인 것이 로짓함수이다. 로짓함수를 사용한 변수가 1개인 일반화 선형모형은 아래와 같이 정의된다. 이를 로지스틱 회귀분석 (logistic regression) 이라고 부른다.
log(μ1−μ)=α+βx
좌측을 살펴보면 log odds 라는 것을 알 수 있다. (=> log(성공확률/실패확률) 이므로) 즉, 로지스틱 회귀분석은 log odds 를 설명변수들의 조합으로 예측하는 것을 의미한다. odds 가 아닌 확률(평균) 의 관점에서 로지스틱 회귀분석은 아래와 같이 써볼 수 있다.
μ=exp(α+βx)1+exp(α+βx)
또한 로지스틱 회귀 분석에서 중요한 것은 beta 값의 해석이다. 만약 x가 연속형인 경우 x+1과 x의 odds 를 구해서 odds ratio 를 구해보자. 위 식에 넣어 계산해보면, OR = exp(beta) 가 나온다. 양변에 log 를 취해주면 log(OR) = beta 라는 것을 알 수 있다.즉, x가 1단위 증가했을 때의 log(OR) 값이 beta 라는 것을 알 수 있다.
한편, Y가 베르누이 분포를 따르는 경우에 사용할 수 있는 다른 연결함수로는 프로빗 연결함수가 있다. 프로빗 연결함수를 사용한 일반화 선형 모형을 프로빗 모형이라고 부른다. 프로빗 모형은 표준정규분포의 누적분포함수의 역함수를 연결함수로 사용한다. 누적분포함수의 역함수를 연결함수로 사용한다는 의미가 무엇일까? 누적분포함수는 0~1사이의 값을 갖는다. 즉, 어떤 -무한대~무한대에 있는 X라고하는 값을 0~1 사이로 변환하는 함수이다. 이에 역함수이기 때문에 0~1사이의 값을 -무한대~무한대로 바꾸어주는 함수가 된다.
도수 자료의 경우에는 일반화 선형모형중 포아송 회귀분석을 해볼 수 있다. 도수 자료란 반응 변수가 도수 (count)로 이루어진 자료를 의미한다 (예를 들어, 교통사고 수, 고장 수 등...). 도수자료는 양의 방향에서만 존재한다. 교통사고수가 마이너스일 수는 없다. 반면, 설명변수의 조합인 체계적 성분은 -무한대~무한대의 범위를 갖는다. 이를 변환하기 위해서, 포아송 회귀분석에서는 연결함수로 log 를 활용하여 좌변이 -무한대~무한대의 값을 갖도록 변환한다. 포아송 회귀분석 식은 아래와 같다.
log(μ)=α+βx
이는 평균의 관점에서는 아래와 같이 쓸 수 있다.
μ=exp(α+βx)
x가 t일 때와 t+1일때의 mu 값을 비교해보자. 위 수식에 대입하면x가 t+1 일 때의 mu 와 t 일 때의 mu 의 ratio 는 exp(beta) 가 됨을 알 수 있다. 즉, 포아송 회귀분석과 같은 log linear regression 에서 beta 를 해석하는 방법은 "x 가 1단위 증가했을 때 Y값의 평균이 exp(beta)배 증가한다." 이다.
포아송 회귀 관련해서는 종종 이런 문제가 발생할 수 있다. 만약, X가 차량 사고수에 미치는 영향을 포아송 회귀로 모델링을 하려고하는데, 지역별로 데이터가 수집 되었고, 지역별로 기본적인 차량의 개수가 달라 사고수가 이에 영향을 받는다고 해보자. 이 때, "사고율" 을 반응 변수로해서 모델링할 수 있다. 차량의 개수를 t라고 하자.
log(μ/t)=α+βx
사고수의 관점에서 아래와 같은 수식으로 변환할 수 있다. 이 때, log(t) 를 offset 이라고 한다.