유전학 분야에서 딥러닝의 발전은 정밀 의학(personalized medicine) 에 구체적으로 어떻게 기여할 수 있을까?
1. 질병에 영향을 주는 유전적 변이 찾기 : 정밀 의학의 한가지 목적은 개인의 질병에 대한 위험도를 정밀하게 추정함으로써, 질병의 조기 발견 및 예방을 하고자하는 것이다. 그리고 그 중심에 있는 것이 과거엔 분석이 어려웠던 유전 정보라고 할 수 있다. 딥러닝 모델은 대규모 유전 데이터에서 유의미한 연관성을 발견하는 것에 기여한다. 예를 들어, 딥러닝을 활용하면 유전자 변이와 특정 질병 간의 관계를 더욱 잘 파악할 수 있다. 더욱 잘 파악한다는 것은 무슨 의미일까? 대표적으로 유전적 변이간의 교호작용 (interaction) 을 예로 들 수 있다. 교호작용이란 쉽게 말해 '시너지' 이다. 에를 들어, A 라는 유전변이가 질병 위험도에 3만큼 기여하고, B 라는 유전변이가 질병 위험도에 5만큼 기여한다고 하자. A,B변이가 모두 있는 사람이 질병 위험도가 30이 증가한다고 하면 기대치 8보다 22높은 값이다. 이런 경우 유전적 변이간에 교호작용 (gene-gene interaction) 이 있다고 한다. 일반적인 통계적인 방법으로도 이를 찾을 수 있지만, 경우의 수가 너무 많아 computational cost 도 크며, 실제 존재하는 interaction 을 잘 찾아내지 못할 가능성 (낮은 statistical power) 도 높다고 알려져 있다.
딥러닝은 이러한 interaction 을 detection 하는데 더 효율적이라고 알려져 있다. 따라서, 개인의 유전 정보 기반 질병의 위험도 평가를 더욱 정확하게 할 수 있고, 이는 질병의 조기 발견 및 예방에 기여할 수 있다. 참고로, 딥러닝에서 유전자 변이와 질병간의 연관성을 파악할 때는, SNP 데이터에 feature engineering 방법 (예를 들면, PCA) 등을 적용해 차원 축소를 하고, 모델의 input 으로 넣는 방법이 많이 사용된다.
2.DNA 의 전사 (Gene expression) 에 영향을 주는 유전적 변이 찾기: 어떠한 유전자 변이가 질병에 영향을 주는 대표적인 경로는 유전자 변이가 유전자 발현(gene expression)에 영향을 주고, 이 유전자 발현의 영향이 질병에 영향을 주는 것이다. 이에, 반응변수(y) 를 질병이 아닌 gene expression 등으로 두고, gene expression 에 영향을 주는 유전자 변이를 찾는 연구가 많이 이루어지고 있다. 보통 coding-variant 의 경우 해당 variant 가 gene expression 에 영향을 준다는 것을 비교적 쉽게 파악할 수 있다. 그러나 문제는 genome 에 대부분을 차지하는 non-coding region 에 위치한 variant 라고 할 수 있다. 딥러닝을 통해 non-coding variant 에 대한 정보(annotation) 을 쌓아, 이를 GWAS 의 결과를 해석하는데 사용할 수 있다.
보통 질병에 영향을 주는 유전적 변이를 찾는 과정에서는 SNP array 등을 많이 사용하는데, gene expression 에 영향을 주는 변이를 딥러닝을 통해 찾는 과정에서는 sequence data (ATGC.... 와 같은) 를 직접적으로 input 으로 넣는 경우가 많다. SNP array 를 사용했을 때와 비교하여 sequence data 를 사용하는 경우, 정보의 손실 (insertion/deletion 등)이 적기 때문일 것이다. 이는 질병에 인과적인 영향을 주는 causal variant 를 찾는 과정에 도움을 주기 때문에 유전적 리스크를 평가하는데 도움을 줌으로써 정밀 의학에 기여할 수 있다.
3. 약물 반응 예측: 정밀 의학의 다른 목표 중 하나는 맞춤형 약물이라고 할 수 있다. 어떤 사람 A 에게는 잘 듣는 약물이 B 라는 사람에게는 잘 안들을 수 있다. 지금까지는 '평균적으로 잘 working 하는 약물' 을 모든 환자에게 투약하는 방식으로 치료 등이 이루어졌다면, 정밀의학 시대에서는 개인에게 잘 맞는 약물을 투약하는 것이 목표라고 할 수 있다. 딥러닝은 환자의 유전적 프로파일을 바탕으로 약물 반응성을 예측할 수 있다 따라서 특정 약물에 대한 환자의 반응을 예측하고, 부작용의 가능성을 최소화하는 데 도움을 줄 수 있다. 이 때의 input 은 genetic data (SNP array, sequence) 등이 될 것이다. 반응변수y는 약물 반응성이 될 것이다. 방법론적 측면에서 보자면 '질병 위험도 예측' 과 '약물 반응성 예측' 은 거의 비슷하다고 볼 수 있을 것이다.
LD score regression 은 Genome-wide association study(GWAS) 에서 특정 trait 의 polegenicity 를 추정하기 위해 사용하는 방법이다. LD score regression 은 GWAS summary statatistics 를 기반으로 SNP-heritability 추정, SNP-heritability 기반 genetic correlation 의 추정 등 다양한 measure 들을 계산하는데에 활용되고 있다. 본 문서에서는 LD score regression 의 등장 배경과 의미에 대해 알아보고자 한다.
polygenic trait 에 대해 GWAS 를 수행한 후에, 각 SNP 들의 p-value 의 분포를 살펴보면 null distribution 과 비교하여 값들이 낮게 나타나는 것을 확인할 수 있다. 이렇게 높게 나타나는 검정 통계량은 형질이 polygenic 함을 의미할 수도 있지만, confounding bias 나 population stratification 가 영향을 주었을 수도 있다. polygenicity 로부터 위와 같은 bias 를 분리해내는 방법이 LD score regression 이며, LD score regression 은 이 과정에서 Linkage Disequillibrium (LD) 와 검정 통계량 (test statistics) 의 관계를 이용한다.
"Both polygenicity (i.e. many small genetic effects) and confounding biases, such as cryptic relatedness and population stratification, can yield inflated distributions of test statistics in genome-wide association studies (GWAS). "
LD score regression 의 아이디어
어떤 SNP j 에 대해서 이 LD 관계에 있는 SNP 들이 많을 수록, polygenic 한 trait 에 대해서는 test statistics 이 높게 나올 가능성이 높다. LD 는 유전자 변이간의 연관성을 의미한다. 만약, 어떤 SNP 이 LD 관계에 있는 SNP 이 많다고 하면, causal variant 와 LD 관계일 가능성이 높고, causal variant 와 LD 관계라면, test statistics 가 높게 나온다. 따라서 LD 관계에 있는 SNP 이 많으면, test statistics 가 높게 나올 가능성이 높다. LD score regression 은 이렇게 LD 관계에 있는 SNP 이 많은 SNP 일 수록 test stat 이 높게 나올 가능성이 높다라는 관계를 이용하는 방법이다. 그리고, 이러한 경향성이 강한 trait 일 수록 polygenicity 가 강하다고 말할 수 있다. (즉, LD score 와 test stat 의 연관성이 강할 수록 polygenic effect 로 phenotype 을 설명할 수 있는 비중이 높다.)
GWAS test statistics
LD score regression 에서는 chi-square value 를 regression 의 종속변수로 선정한다. chi-square value 는 무엇일까? 일반적으로 GWAS 결과로 effect size (beta) 와 standard deviation (sd) 값이 나오게 된다. 이 때, beta/sd 를 z-value 라고 한다. (이는 beta = 0 이라는 귀무가설 하에 구한 z-score 이다.) z-value 관측된 beta 값이 0으로부터 몇 standard deviation 떨어져 있는지를 의미한다. 이 때 chi-square value 는 z value 의 제곱으로 계산된다. z-value 는 표준정규분포를 따르며, 표준 정규 분포의 확률변수 z 의 제곱은 자유도가 1인 chi-square 분포를 따르기 때문이다.
LD 와 LD score 는 무엇일까?
먼저 LD 와 LD score 를 계산하는 방법을 간단히 알아보자. 일반적으로 두개의 SNP A,B 의 LD 와 관련된 지표 D 와 r^2 은 아래와 같이 계산된다. D 값이 높을 수록 A,B 변이는 함께 나타날 가능성이 높음을 의미 한다. 만약, P(A)=0.3, P(B)=0.4, P(A,B) = 0.15 라고 하면, D는 0.03 으로 계산되며, A,B 는 LD 관계가 아닐 것으로 판단된다.
특정 SNP j 에 대한 LD score 는 아래와 같이 계산된다. LD score 는 각각의 SNP 에 대해 다른 모든 SNP 들과의 LD 값 (r^2) 들을 더한 값으로 볼 수 있다.
$$ l_j = 1 + \sum_{k \neq j}r_{jk}^2 $$
LD score 와 test statistics (chi square value) 의 관계를 아래와 같이 시각화해볼 수 있다. 아래 차트는 LD score 의 bin 과 평균 chi-square value 의 관계를 보여준다. 직선은 아래 점들을 대상으로 단순 선형 회귀 분석을 한 결과를 표현한다 (아래 차트에서 각 점들에 해당 하는 SNP 의 갯수에 가중치를 두어 regression 을 돌리면 결국 전체 snp 과 chi-square 를 대상으로 regression 을 돌린 것과 같은 값이 나오게 될 것이다).
LD score 와 test stat (chi-square value) 의 관계 및 선형 회귀 분석의 결과
이를 LD score regression 이라고 하며, 이 선형 회귀 분석에서 기울기는 polygenicity 를 반영하고, 절편은 bias 를 반영한다. LD 와 chi-square 의 연관성 (기울기) 이 polygenicity 이며, 전반적으로 chi-square 가 inflation 이 된 정도 (절편) 가 bias 라는 것이다. 이러한 방법을 통해 polygenicity 와 bias 를 분해할 수 있게 된다.
또한, LD score regression 에서 기울기는 heritability 를 반영한다. 아래 그림과 같이 heritability 가 높을 수록 LD score regression 의 기울기가 커지게 된다.
다양한 heritability 값들에 대한 LD score regression slope 와의 관계 (simluated data)
또한, 기울기는 샘플 사이즈와 사용한 SNP의 전체 개수에도 영향을 받는다. 샘플 사이즈 N 이 커질 수록 chi-square 값이 커지고, 사용한 SNP 의 개수가 많아질 수록, LD score 의 값이 기본적으로 높아진다. 이를 고려하여, 특정 SNP j 의 test stat 을 설명하는 아래와 같은 regression 모델을 고려할 수 있다.
$$ E[\chi^2 | l_j] = Nh^2/M l_j + Na + 1 $$
기울기는 heritability 와 N, M 으로 분해하여 나타낸다. 또한 절편은 Na + 1 로 표현되는데, 이 때, a 가 population structure 또는 confounding bias 와 같은 요인으로 인해 test stat 이 inflation 된 정도를 의미한다. 1이 더해진 이유는, LD score 가 0 인 SNP (즉, 그 어떤 SNP 과도 LD 관계에 있지 않은 SNP) 의 경우, chi-square value 는 causal variant 가 아닌 이상 기댓값은 1일 것이다 (자유도가 1인 chi-square distribution 의 평균값). 따라서, 절편은 1에 가까울 것이며, 1에서 벗어난 만큼을 bias 로 판단하겠다는 의미를 가진다.
Bivariate LD score regression
두개의 trait X, Y 에 대한 test statistics 를 이용해 LD score regression 을 하는 것을 Bivariate LD score regression 이라고 한다. 구체적으로, 두개의 trait X, Y 에 대한 각각의 z-value 의 곱에 대하여 LD score regression 을 한다. 앞선 LD score regression 에 대해서는 chi-square value 를 사용했는데, chi-square value 는 z-value 의 곱이다. 즉, z^2 대신에 z_x * z_y 를 넣어서 regression 을 한다는 것이다.
참고) X_n 이 표준정규분포로부터 추출된 random variable 일 때, X_n 의 제곱의 합은 자유도가 n인 chi-square 분포를 따른다.
위에서는 자유도가 1인 chi-square value 이기 때문에 z^2 = chi-square 가 된다.
$$ X^2_1 + X^2_2 + ... X^2_n \sim \chi^2(n) $$
이 때의 기울기는 무슨 의미를 가질까? 만약 두개의 z-value 간에 아무런 연관성이 없다면, 1을 중심으로 퍼져있는 분포를 나타내게 된다. (z^2 의 분포는 자유도가 1인 카이제곱 분포를 따르며, 자유도가 1인 카이제곱 분포의 기댓값은 1이기 때문) 하지만, z-value 간에 연관성이 있다면, 기울기를 갖게 되며, 이 때의 기울기는 두 trait 간의 유전적 연관성을 의미한다. 두 trait 간에 유전적 연관성을 나타내는 지표로 co-heritability 가 있다. 기울기는 co-heritability 를 반영한다고 볼 수 있다. 이를 모델링하면 아래와 같이 표현할 수 있다.
genetic score, heritability, co-heritability, genetic correlation 관련 개념 정리
phenotype variance 의 분해. 본 문서에서는 , G 와 E만 고려한다.
phenotype Y 의 분산은 genetics 로 설명되는 분산과 environment 로 설명되는 분산으로 나누어진다. 유전율 (heritability) 는 phenotype(또는 trait) 의 분산에서 genetics 를 통해 설명되는 분산을 의미한다. heritability 는 0~1 사이의 값을 가진다.
$$ Var(Y) = Var(G) + Var(E) $$
$$ h^2 = Var(G)/Var(Y) $$
genetics 로 설명되는 분산이란 무엇일까? phenotype 을 예측하기 위해 유전정보를 이용해 어떠한 score 를 만들고, 이를 genetic score 라고 하자. genetic score 는 phenotype 에 대한 예측 값이며, 이 값이 높을 수록 phenotype 의 값이 높을 가능성이 높음을 의미한다 (만약 질병과 같은 binary trait 인 경우, 질병의 걸릴 가능성이 높음을 의미한다.)
genetic score 는 유전체 정보를 이용해 구한 phenotype Y 에 대한 예측값이다. 따라서 아래와 같이 쓸 수 있는데 heritability 의 식이 결정계수의 식과 닮아 있음을 알 수 있다. 결정계수는 전체 분산중 어떠한 모델을 통하 예측값의 분산이 차지하는 비율이며, 이것이 곧, 모델을 통해 설명되는 분산을 의미한다.
$$ h^2 = Var(\hat{Y}) / Var(Y) = r^2 $$
genetics 로 설명되는 분산은 genetic score 의 분산으로 정의할 수 있다. 만약 genetic score 를 구할 때, additive genetic effect 만 고려하여, additive genetic score 를 구해 heritability 를 구한 것을 narrow-sense heritability 라고 한다.
$$ h_n^2 = Var(AG)/Var(Y) $$
만약 , Y 가 standardization 이 되어 있다고하면, Y의 평균은 0이고, Y의 분산은 1이다. 그러면, 간단히, additive genetic score 의 분산이 바로 narrow sense heritability 가 된다.
"If the traits are standardized (that is, phenotypic variance = 1) and the genetic values consider only the additive genetic effects, then the genetic variances are narrow-sense heritabilities."
$$ h_n^2 = Var(AG) $$
두 가지 trait 의 유전적인 연관성을 정량적으로 표현하는 지표로 coheritability 라는 개념이 있다.
"Co-heritability is an important concept that characterizes the genetic associations within pairs of quantitative traits."
co-heritability 는 아래와 같이 정의되며, -1~1 사이의 값을 가진다.
$$ h_{x,y} = \frac{Cov(g_x,g_y)}{Var(X)Var(Y)} $$
이 식의 의미를 살펴보면 분자의 covariance 에 Cov(X,Y) 가 오게 된다면, pearson 상관계수와 같음을 알 수 있다. 이 식은 Cov(X,Y) 대신에 X,Y 에 대한 genetic score 를 대입시킴으로써, 두 trait 의 유전적 상관성을 표현했다고 볼 수 있다. 여기서도 마찬가지로 trait X,Y 를 평균이 0이고 분산이 1인 표준화된 trait 을 사용했다면, Var(X) = Var(Y) = 1 이기 때문에 아래와 같다.
$$ h_{x,y} = Cov(g_x,g_y) $$
두 가지 trait 의 유전적인 연관성을 정량적으로 표현하는 지표로 genetic correlation 이라는 개념도 있다. genetic correlation 은 아래와 같이 정의된다.
"The genetic correlation is a quantitative genetic parameter that describes the genetic relationship between two traits"
위 식은 pearson 상관계수의 식과 같으며, genetic correlation 의 통계적인 의미는 X,Y 의 genetic score 상관성 (pearson 상관계수) 라고도 할 수 있다. 만약 두가지 trait, 예를 들어 키와 발가락 길이의 유전적 연관성이 높다라고 한다면, 유전자를 통해 예측한 키 (키에 대한 genetic score) 와, 예측된 발가락 길이 (발가락 길이의 genetic score) 의 연관성이 높을 것이다. 이를 수치화한 것이 genetic correlation 이라고 볼 수 있다. genetic correlation 도 마찬가지로 -1~1사이의 값을 가진다.
genetic correlation 과 co-heritability 모두, 두가지 trait 의 유전적 연관성을 표현한다. 둘의 차이점은 무엇일까? trait X,Y 가 표준화 되어있다고 하면 genetic correlation 은 아래와 같이 정의된다. 아래 식을 보면, genetic correlation 은 co-heritability 가 X,Y 각각의 trait 의 heritability 로 보정된 식임을 알 수 있다.
$$ \rho_g = \frac{h_{x,y}}{\sqrt{h^2_x h^2_y}} $$
따라서, 두 trait 의 heritabilty 값이 작더라도, genetic correlation 은 높을 수 있다. 예를 들어, 발가락 길이와 키의 heritability 가 10% 라고 하자 (실제로는 더 높을 것이나 예시임). 즉, 전체 분산에서 genetic score 의 분산이 차지하는 부분이 10% 이다. 하지만, 두개의 genetic score 의 연관성이 높다라고 하면, genetic correlation 은 높게 추정될 수 있다. 따라서, genetic correlation 을 해석할 때, trait 을 genetics 가 설명하는 비중 (heritability) 도 함께 고려해야할 필요가 있다.
참고자료
- Genetic correlations of polygenic disease traits: from theory to practice, Nature review genetics, 2020
- Optimal Estimation of Co-heritability in High-dimensional Linear Models
- Statistical methods for SNP heritability estimation and partition: A review
DNA (deoxyribonucleic acid) 의 구조와 유전암호 (genetic code, codon) 이 발견된 이후 수십년간, 인간 유전학 연구는 단백질 코딩 유전자 (protein-coding gene) 의 기능과 구조를 이해하고 왜 단백 코딩 유전자에 변이가 생겼을 때, 질병이 발생하는지에 대한 연구가 중점적으로 이루어져 왔습니다. Central dogma 라고 불리는 생물학의 중심 원리는 유전자가 mRNA 로 전사 (transcribe), 다시 mRNA는 단백질로 번역(translate) 된다고 상정하고 있습니다. 직관적인 유전암호 덕분에 단백질 코딩 유전자에 변이가 생겼을 때, 최종 산물인 단백질의 아미노산 구성에 어떤 영향이 미칠지 쉽게 예측할 수 있었습니다.
멘델리안 질병
가족 직접성 (Familiar aggregation) 을 강하게 보이고, 가족 내에서 질병이 예측할 수 있는 패턴으로 관찰되는 멘델리안 질병 (Mendelian disease)은 한 유전자의 돌연변이가 생겨 발생합니다.1983년 헌팅턴 질병의 유전적원인을 찾은 것을 시작으로, 다한 멘델리안 질병의 인과성이 있는 유전적 변이를 positional cloning 방법을 통해 잇달아 발견했습니다. 이를 통해 멘델리안 질병에 대한 유전적 원인을 어느정도 이해할 수 있었습니다.
복합 질환과 전장 유전체 분석
하지만 현재 흔하고, 질병 부담이 큰 질병, 예를 들어 심혈관 질환 (cardiovascular disease), 암 (cancer), 알츠하이머 병 (Alzheeimer's disease), 파킨슨 병 (Parkinson's disease), 당뇨병 (type 2 diabetes) 등의 질병의 경우, 하나의 유전자의 돌연변이로 인해 발생하지 않습니다. 이러한 질병을 "복합질환 (complex trait)" 라고 하는데, 복합질환은 여러 유전 요인 및 환경 요인과 그들의 조합에 의해 영향을 받아 발생합니다.
복합질환과 연관성이 있는 DNA 의 돌연변이 (genetic variant) 를 찾기 위해 전장 유전체 분석 (genome-wide association study, GWAS) 이 2005 년부터 시작됩니다. 최초의 GWAS 연구라고 불리는 연구는 2005년 science에 출간된 나이 관련 황반병성 관련 연구입니다.
Complement Factor H Polymorphism in Age-Related Macular Degeneration, Science, 2015)
complement factor H 유전자 주위의 유전적 변이를 나이관련 황반 변성과 연관시킨 이 연구를 시작으로해서 전세계 수많은 연구자들이 복합질환과 연관성이 있는 유전적 변이를 찾기 위한 수많은 전장 유전체 분석 연구를 수행하였습니다. 전장유전체분석은 통계적으로 유의하게 질병과 연관성이 있는 유전적 변이를 찾는 방법이며, 일반적으로 단일염기 다형성 (Single nucleotide polymorphism) 이 많이 사용됩니다. 같은 질병을 대상으로한 GWAS 연구에서 반복적으로 통계적으로 유의하다고 발견되면, 이 변이는 실제 연관성이 있는 (질병의 위험도를 높이는) 변이라고 생각해볼 수 있었습니다.
하지만 문제는, GWAS 연구의 결과로 발견된 변이 (GWAS Hit) 라도 그것이 실제 생물학적으로 질병의 위험도를 높이는 변이가 아닐 수 있다는 것입니다. 어떤 변이가 질병과 연관성이 있다는 사실은, 해당 인구집단 내에서 개인의 질병 위험도를 계산하는데에는 유용하게 쓰일 수는 있어도, 이것을 통해 질병의 생물학적인 메커니즘을 이해할 수 있는 것은 아니었습니다. 이유는 다음과 같습니다.
1) 많은 GWAS Hit 들이 실제 연관성이 있는 변이 (causal variant) 와 Linkage disequilibrium 관계에 있음
2) 많은 GWAS Hit 들이 non-coding region 에 위치 (> 90%)해 있는데 이 지역이 무엇을 하는지 모름
1) Linkage disequilibrium 이란 genome 상의 특정 부분의 서열 (genotype) 이 다른 genotype 과 연관성이 있는 것을 말합니다. LD 가 있는 것은 두 genotype 을 골랐을 때, random 하게 나오는 쌍의 빈도보다 얼마다 deviation 되어있는지를 통해 판단하며, LD 는 genome 상의 실제 거리가 가까울 수록 높습니다. 따라서 causal variant 과 LD관계에 있는 변이들이 GWAS hit 으로 나오게 되는 것입니다. 만약 genome 상의 X 라는 위치에 AA, Aa, aa 3개의 genotype 이 있을 수 있는데, a가 causal variant 라고 할 때, X와 LD 관계에 있는 Y 에 b 라고 하는 대립유전자가 a와 연관이 되어있으면, b도 GWAS hit 으로 나올 가능성이 큽니다. 그리고 GWAS 자체가 imputation 이라는 방법을 이용해서 LD 를 '이용' 해서 통계적으로 유의한 variant 를 찾아내기도 합니다.
2) 90 % 이상의 GWAS hit 들이 non-coding region 에 위치해 있습니다. 즉, genetic code를 이용해서 해당 변이가 어떤 결과를 불러오는지 알아낼 수 있는 protein coding region 에 비해 non-coding region 은 이러한 해석이 불가능했습니다. 한 가지 가능한 해석은 이 지역이 유전자 근처에 위치해 유전자 발현에 영향을 주는 지역 (cis-regulatory region, cRE) 이라는 것입니다. 이것이 가능한 해석이긴 했지만, 진핵생물의 경우, 전사 조절 (transcriptional regulation) 이 워낙 복잡하기 때문에, 그것이 LD 인지, cRE 인지 알기가 힘들었습니다. 유전자 발현은 조직별로 다르게 나타나며, 어떠한 variant가 transcription 에 영향을 주는 경로는, DNA methylation, histone modification, splicing, transcription factor binding intensity change, mRNA stability 등으로 매우 다양합니다.
Functional genomics
Functional genomics의 최종적인 목적은 genome 상의 element 들이 어떤 기능을 하는지 알아내고자 하는 것입니다. GWAS hit 들의 많은 부분이 eQTL 과 겹칩니다. 하지만 문제는, variant가 expression 을 '아주 조금' 변화시킨다는 것입니다. 대부분의 variant 들인 target gene 의 expression을 평균적으로 2배 미만으로 증가시킵니다. 그리고 왜 expression 에 영향을 주는지 확실하게 밝히기가 어렵습니다. 현재까지로는, 복합질환의 경우 variant가 최종 표현형 (phenotype)인 질병에 영향을 주는 메커니즘이 수많은 variant 가 target gene의 expression 에 조금씩 영향을 주고, 이것이 최종적으로 질병 발생의 위험도를 증가시키는 것으로 이해할 수 있습니다. functional genomics 에는 많은 분야가 있지만, 아래 두 관계에 대한 생물학적인 이해를 하고자하는 것이 중요해보입니다.
1) variant -> target gene expression
2) target gene -> phenotype
genotype-phenotype 관계를 생물학적 기능을 이해함으로서 풀고하는 분야가 바로 functional genomics 라고할 수 있습니다. 이를 위해 다양한 실험 데이터 (chip-seq, 5c, hi-c, dnase-seq ...)와 생물정보학적 방법이 동원되고 있습니다. functional genomics 의 한가지 특징은 전통적인 'gene-by-gene 분석보다는 NGS 등을 이용한 genome-wide 분석이 장려된다는 것입니다.
Functional genomics (Wikipedia, Sep, 2019)
Functional genomics is a field of molecular biology that attempts to describe gene (and protein) functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects (such as genome sequencing projects and RNA sequencing). Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional “gene-by-gene” approach.
Functional genomics 의 대표적인 데이터베이스
GTEX (Genotype-Tissue Expression): GTEX 는 genotype 과 tissue specific gene expression 을 저장하고 있는 DB 입니다. 50 개 이상의 tissue 에 대한 gene expression level 과 genotype 데이터를 갖고 있습니다. 이 때 어떤 genotype 이 어떤 tissue 의 어떤 gene 의 expression 에 영향을 주는 것이 통계적으로 관찰되면 이를 eQTL (expression quantitative trait loci) 라고 부릅니다. 실제로 많은 GWAS hit 들이 eQTL 과 겹치는 것으로 나타납니다 (ASHG, 2018).
ENCODE (Encyclopedia of DNA Elements): genome 상에서의 transcription, transcription factor association, chromatin structure, histone modification을 밝혀내기 위한 프로젝트입니다. 이러한 genome 상에서의 기능적인 요소들을 식별함으로써 현재 인간 genome의 80%의 부분이 최소 1개 이상의 biochemical function 을 한다고 생각되어지고 있습니다.
genome 상의 non-coding region 의 functional element 를 찾기 위한 ENCODE 프로젝트
본 포스팅은 부모-자식 데이터를 통해 heritability를 추정하는 것을 포스팅한다. 아래 데이터는 parent와 child의 height를 나타내는 수치이다. 옥수수의 키라고 생각하자. 알고 싶은 것은 child의 키가 유전적 요소로 설명되는 정도이다. 우리는 이 수치를 heritability(h^2)라는 값으로 표현한다. 데이터는 이곳에서 다운로드 할 수 있다.
\[ h^2 = \frac{V(G)}{V(Y)}\]
library(data.table)
## Warning: package 'data.table' was built under R version 3.4.3
##
## Call:
## lm(formula = Galton$child ~ Galton$parent)
##
## Residuals:
## Min 1Q Median 3Q Max
## -7.8050 -1.3661 0.0487 1.6339 5.9264
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 36.87187 1.98827 18.55 <2e-16 ***
## Galton$parent 0.45700 0.02909 15.71 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 2.239 on 926 degrees of freedom
## Multiple R-squared: 0.2105, Adjusted R-squared: 0.2096
## F-statistic: 246.8 on 1 and 926 DF, p-value: < 2.2e-16
회귀계수가 0.457이다. 이 때, 유전율은 0.457*2 = 약 92%로 추정된다. 왜 그런지 살펴보기 위해 우선 beta의 추정치를 아래와 같이 회귀식의 양변에 Cov(, X)를 취해 구해보자.
\[ Y = \alpha + \beta X \]\[ Cov(Y, X) = \beta Cov(X,X) \]\[ \hat{\beta} = \frac{Cov(Y,X)}{Var(X)} \]
Phenotype Y에 대해 아래와 같은 일반적인 Phenotype에 대한 모델을 세우자. 이 때, A,D,C,E 각각의 컴포넌트들에 대한 것은 나중에 살펴보고 i, j 두 개체의 Covariance만 살펴본다. A : additive effect D : dominance effect C : shared environment effect E : residuals \[ Y = A + D + C + E\] r은 relatedness, k2는 genome상의 IBD2의 비율, a는 shared environment를 나타내는 수치이다. 만약 shared environment이면 a=1, 아니면 a=0이다.
이 Covariance 모델은 어디까지나 가정이다. 이 가정 하에서 각각의 Variance를 추정하고 이를 통해 heritability를 추정하게 된다.
이제 현재 데이터에서 위 식을 적용하자. 이 때, r=0.5, k2=0, a=0을 가정하자. (부모-자식 간의 IBD2=0이므로 k2=0이다. ) 그러면 랜덤으로 i번째 부모-자식 pair를 골랐을 때, Covariance 모델은 아래와 같다. (P : Parent, C : Child의 약자)
이러면 Var(D)가 사라지기 때문에 우리는 쉽게 Var(A)를 데이터를 통해 추정할 수 있다.
<solar-base> is the directory name where the bulk of this release of SOLAR
is to be installed. To keep it separate from other releases, the release
number should be included in this directory name. For example, we use:
/opt/appl/solar/8.1.1
<solar-script-dir> is the existing directory to which the 'solar' script
that starts SOLAR is to be put. /usr/local/bin is a typical choice.
This should be in the PATH of everyone who will use solar.
You can move this script later if desired. If you already have an older
script named 'solar' there, you must rename or delete it first.
이 때 solar-base에는 실제 실행 파일이 위치한 곳, solar-script-dir에는 실행파일을 연결할 파일의 경로를 써준다. 일반적으로 solar-script-dir은 /usr/local/bin 파일에 위치하도록 하고, solar-base는 아무 곳에나 놔두어도 되지만 /usr/local/bin에 위치해도 크게 상관은 없다.
인간의 DNA 속 기능의 단위인 유전자는 단백질을 암호화하고 있고, 유전자가 암호화하고 있는 단백질은 세포의 기능을 결정합니다. 따라서 어떤 세포 안에서 생성되는 수천개의 유전자는 그 세포가 무엇을 할지를 결정합니다. 각각의 세포는 다른 유전자를 생성함으로써 다른 기능을 수행하는 것입니다. 유전자 발현이란 이렇게 DNA가 최종 생산물인 단백질(혹은 ncRNA)을 생성하는 과정을 뜻합니다. 그리고 유전자 발현은 각각의 세포마다 다르게 일어납니다. 예를 들어, 시세포, 지방세포, 뇌세포 등은 각각 다른 단백질을 생성함으로써 원하는 생물학적 기능을 수행하게 됩니다. Central Dogma라고 불리는 DNA가 전사 되어 RNA가 되고 RNA가 번역되어 단백질이 되는 각각의 단계는 생성되는 단백질의 종류와 양을 조절할 수 있는 조절 포인트입니다.
어떻게 유전자 발현이 조절될까?
이처럼 유전자가 단백질이 되어가는 과정 속에서 유전자의 발현을 조절하여 유전자의 산물(전사체 혹은 단백질)의 양을 조절하는 것을 유전자 조절(gene regulation)이라고 합니다. 세포 내에서의 각 유전자들의 발현량 그 세포의 기능을 결정합니다. 유전자 조절은 여러 단계가 있지만, 그 중 전사 조절이 가장 중요하고 대부분의 유전자 조절이 일어나는 단계입니다. 사실 진핵 생물의 유전자 발현 조절은 매우 복잡합니다. 진핵생물에서는 전사체가 핵을 떠나기 전에 변형이 일어납니다. 단백질을 코딩하지 않는 부분인 "인트론"이 사라지며, 단백질을 코딩하는 부분인 "엑손" 만 남게 됩니다. 이 엑손이 서로 붙게되어 mature mRNA가 생성됩니다. 또 양 끝부분에도 변형이 일어나는데 이는 안정성에 영향을 끼칩니다. 이러한 RNA Processing의 과정 속에서도 유전자 조절이 일어날 수 있습니다. 또는 번역 단계에서도 miRNA 등의 small RNA에 의해 유전자 조절이 이루어지기도 합니다.
Figure 1: 진핵생물에서 DNA가 단백질이 되어가는 과정
DNA의 번역, 비번역 부위가 mRNA로 전사된다.
mRNA 프로세싱 도중 인트론 부분이 제거된다. 엑손 부분만 남아서 연결되며, 양 끝에 특별한 시퀀스가 붙는다. 이러한 프로세싱이 완료되면 mRNA는 핵을 빠져나가 세포질로 가게된다. 일단 세포질로 가게되면 mRNA는 단백질을 생성할 준비가 완료된다.
어떻게 세포는 자신들이 필요한 유전자를 발현 시킬까?
위에서는 유전자가 단백질로 변화하는 과정 속에서 유전자 조절이 언제 이루어지는지 알아보았습니다. 그렇다면 구체적으로 어떻게 세포는 원하는 유전자만 발현시켜 원하는 기능을 수행할 수 있을까요? 이는 각각의 세포들이 제각기 다른 전사 조절 인자들을 갖고 있기 때문입니다. 이러한 조절 인자들은 전사를 촉진(activate)시키기도 하고 방해(repress)하기도 합니다. 전사 조절 인자는 단백질이며, 이 전사조절인자도 유전자들이 코딩하고 있습니다.
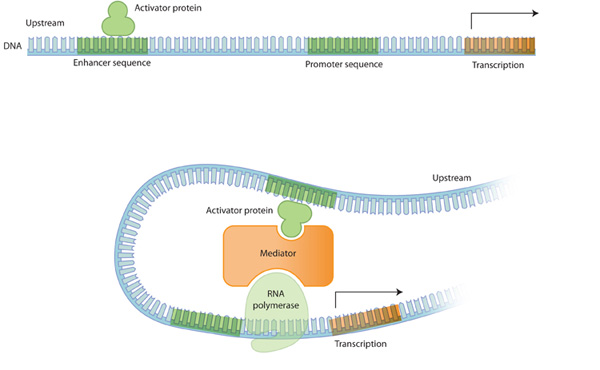
일반적으로 전사는 RNA 중합효소가 소위말하는 프로모터시퀀스에 결합함으로써 시작됩니다. 이 시퀀스는 전사 시작 지점에 근접한 upstream 방향에 존재합니다. (5' 쪽 방향) 하지만 downstream 방향에도 존재할 수 있습니다. (3' 쪽 방향) 비교적 최근에 연구자들은 인핸서 시퀀스라는 것을 발견하였습니다. 인핸서는 전사 조절 단백질들이 결합할 수 있는 결합 위치(binding site)를 제공함으로써 전사에서 중요한 역할을 하는 시퀀스입니다. 이 인핸서의 전사 조절 단백질이 붙게되면 염색질 구조가 변화되어 RNA 중합효소나 조절 단백질의 결합을 촉진하거나 억제하는 역할을 하게됩니다. 이 때 염색질 구조를 open chromatin structure라고 하는데, 이는 유전자 전사가 활성화 된다는 것과 연관이 있습니다. 반면 염색질의 구조가 빽빽한 경우, 전사가 억제되어 있는 것과 연관이 있습니다.
몇몇 조절 단백질은 다양한 유전자의 전사에 영향을 미칩니다. 이는 전사 조절 단백질 결합 위치 (regulatory protein binding site 혹은 transcription factor binding site라고도 함)가 다양한 곳에 존재하기 때문입니다. 결과적으로 조절 단백질은 다양한 유전자의 걸쳐 다양한 역할을 합니다. 조절 단백질의 역할은 단지 어떤 한 유전자의 발현을 조절하는 것이 아니라 다양한 유전자의 발현을 조절합니다. 이것이 각각의 세포가 한 번에 많은 수의 유전자를 조절할 수 있는 하나의 메커니즘입니다.
Figure 2: 전사의 조절
활성 단백질이 인핸서 시퀀스에 결합하게되면 RNA 중합효소를 활성화 시키는 프로모터 부분에 단백질을 끌어들일 수 있고, 이로 인해 전사가 촉진된다. DNA는 위와 같이 굽어져 활성자 단백질은 RNA 중합효소의 활동을 중재하는 다른 단백질들과의 상호작용을 하게 된다.
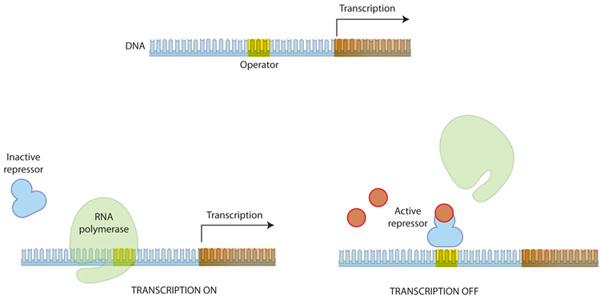
우선 원핵 세포에 대해서 알아보면, 원핵생물에서 조절 단백질은 종종 영양소의 이용가능성에 의해 조절됩니다. 이는 박테리아와 같은 생물이 환경 조건에 반응하여 전사 패턴을 빠르게 조절할 수 있게 합니다. 덧붙여, 원핵생물의 조절 부위는 프로모터와 가깝게 위치합니다.
Figure 3: 프로모터 주위에서의 전사 조절
특정 단백질은 RNA 중합효소에 간섭을 함으로써 전사를 조절한다. 불활성화 상태로 존재하는 억제 단백질(repressor)은 다른 분자에의해 활성화 될 수 있으며, 활성화된 상탱서 operator라 불리는 부위에 결합하여 RNA 중합효소가 프로모터에 결합하는 것을 방해한다. RNA 중합효소가 프로모터 부위에 결합하여야 전사가 개시됨으로 이는 전사를 효과적으로 억제한다.
활성자(activator)는 프로모터 주위에 조절 부위에 결합하여 RNA 중합효소의 활동을 촉진합니다. 억제자(repressor)는 조절부위에 결합하여 RNA 중합효소의 결합을 방해합니다.
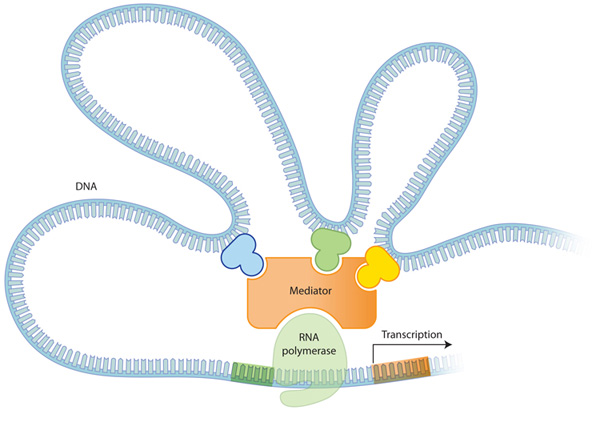
진핵생물의 유전자 발현 조절은 원핵생물에 비해 복잡합니다. 위에서 기술한 것과 마찬가지로 기본적으로는 프로모터 주위 조절 부위에 결합하는 활성자, 억제자에 의해 RNA 중합효소의 활동량이 조절되어, 전사 과정에서의 유전자 조절이 일어납니다. 하지만 진핵생물에서는 그 이상의 많은 수의 조절 단백질이 존재하며, 조절 단백질의 결합 부위는 프로머터와 멀리 떨어져 있는 경우도 많습니다. 이로 인해 유전자 발현의 조절이 더욱 유연하게 됩니다.
Figure 4: 많은 수의 전사 조절 인자
전사 조절 인자들은 각각 다른 역할을 갖고 있다. 위의 3가지 전사 조절인자는 Mediator 복합 단백질과 각각 다르게 상호작용하여 전사를 조절한다. 각각의 조절단백질이 있고 없음, 또한 이들이 어떻게 조합되느냐에 따라 유전자의 발현이 달라진다. 진핵 생물의 유전자 발현의 특징은 이러한 복합한 조절 과정을 통해 같은 유전자라도 다르게 번역될 수 있다는 것이다.
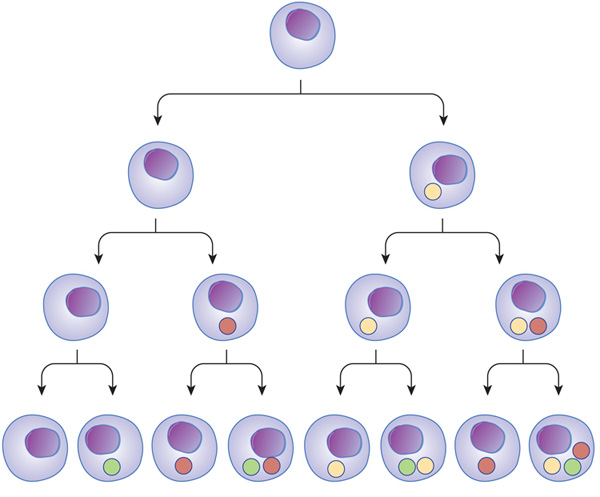
다른 세포 유형은 특징적인 전사 조절인자를 갖고 있습니다. 다세포 생물에서 다른 세포는 각각 다른 조절 인자들의 조합을 갖고 있습니다. 이로인해 각기 다른 기능을 하는 다양한 세포가 생성되고, 기능할 수 있는 것입니다.
Figure 5: 전사 조절인자가 세포 유형을 결정한다.
세포 발달 과정에 따라 전사 조절 인자가 달라지는 것을 나타내는 그림
세포 유형의 다양성은 다른 전사조절인자들의 활동에 의한 것이다.
생명 활동을 위해서 세포는 환경의 변화에 반응해야합니다. 단백질 생산 과정의 중요한 두 가지의 스텝 (전사와 번역)에서의 유전자 조절이 이러한 환경 적응성에 중요한 역할을 합니다. 세포는 필요한 특정 유전자를 발현시켜, 세포의 기능을 수행하며, 또한 환경에 반응하는 유전자 조절을 통해 어떤 유전자가 전사되며 번역될지를 조절합니다.
- 단백질을 코딩하지 않는 DNA 서열이다. * 하지만 miRNA 등의 non-coding RNA를 코딩할 수 있는 것으로 보인다.
3. Transcription Start Site
- RNA 중합효소2(polymerase)가 붙어서 전사가 시작되는 위치이다. (RNA 중합효소2는 12개의 단백질의 중합체로 mRNA를 합성해내는 물질이다.)
4. Promoter
- Core Promoter : TATA 박스(타타 박스라고 읽음)라는 것이 있는데, 이 곳은 실제로 Transcription Factor와 기타 여러 Regulatory Protein이 실제로 붙는 곳이다. 이를 통해 50여개의 단백질 complex가 만들어지며, 이것이 실제로 trascription이 일어나는데 필요한 물질들이다.
- Upstream Promoter : transcription factor 와 기타 조절 단백질(regulatory protein)이 달라붙는 부분이다. Upstream promoter는 gene마다 갯수와 타입이 다르다. transcription factor는 붙어서 해당 유전자를 활성화하거나 비활성화한다.
Promoter는 gene expression을 "총괄" 한다고 보면 되며, 이를 Enhancer가 돕는다고 볼 수 있다. 참고로 잘 알려지지 않은 것 중에 Silencer 라는 것도 있다. 이는 Histon modification과 같은 방식으로 Promoter의 반대 기능 gene expression을 억제하는기능을 수행한다.
5. Enhancer
- 보통 upstream 또는 downstream 방향으로 수천 bp 떨어진 곳에 위치하며, (또는 gene 안에 존재할 수도 있다.) transcription factor가 달라붙게 되면 promoter 쪽으로 굽어서 유전자 발현을 조절한다. 예를 들어, promoter에게 "여기는 brain이니까 이 gene을 더 발현해라!" 라고 말하는 것으로 보면 된다.
유전체에서 유전자 발현 조절 과정 (Regulation)을 수행하는 요소를 찾는 것은 중요한데, 이러한 유전체상의 조절부위(Regulatory region)을 찾는 기본적인 원리는 이 부위에 transcription factor가 달라붙는다는 성질을 이용하여 antibody를 이용한 assay를 하는 것이다.
Linkage mapping/recombination mapping/positional cloning
- rely on known markers (typically SNPs) that are close to the gene
responsible for a disease or trait to segregate with that marker within a
family. Works great for high-penetrance, single gene traits and
diseases.
QTL mapping/interval mapping - for quantitative
traits like height that are polygenic. Same as linkage mapping except
the phenotype is continuous and the markers are put into a scoring
scheme to measure their contribution - i.e. "marker effects" or "allelic
contribution". Big in agriculture.
GWAS/linkage disequilibrium mapping - score
thousands of SNPs at once from a population of unrelated individuals.
Measure association with a disease or trait with the presumption that
some markers are in LD with, or actually are, causative SNPs.
So linkage mapping and QTL mapping are similar in that they rely on
Mendelian inheritance to isolate loci.QTL mapping and GWAS are similar
in that they typically measure association in terms of log-odds along a
genetic or physical map and do not assume one gene or locus is
responsible.And finally, linkage mapping and GWAS are both concerned
with categorical traits and diseases.
Linkage Study : 가족데이터 - 멘델 유전 이용, 알려진 마커로 high-penetrance, single gene disease 멘델리안 질병에 대해서 연구
특정 조건 안에서 allele frequency와 genotype frequency는 영속적으로 보존된다.
위 그림과 같이 allele frequency가 freq(A) = p, freq(a) = q일 때, 다음 세대의 genotype frequency의 기댓값은 freq(AA) = p^2, freq(Aa)=2pq, freq(aa)=q^2 이다. 이 genotype frequency를 통해 다시 allele frequency를 계산하면 freq(A)=p, freq(a)=q가 나온다. 즉, allele frequency와 genotyep frequency는 계속해서 유지된다.
allele frequency와 genotype frequency에서 중요한점
1. genotype frequency를 알면 allele frequency를 알 수 있다.
2. allele frequency를 알아도 genotype frequency를 반드시 알 수는 없다.
2번의 경우 예를 들어, freq(A) = 0.5, freq(a) =0.5 라하자. 그러면 하디 바인베르크 법칙을 만족하면 genotype frequency는 freq(AA)=0.25, freq(Aa)=0.5, freq(aa)=0.25일 것이다 하지만 freq(AA)=0.5, freq(Aa)=0, freq(aa)=0.5 여도 주어진 조건에 맞는다. 따라서 allele frequency를 알아도 genotype frequency를 반드시 알 수 있는 것은 아니다.allele frequency는 재료이다. 그것이 어떻게 조합되어 genotype을 구성할지는 확정적이지 않다. 이는 하디 바인베르크 법칙에 위배된다.
그렇다면 하디 바인베르크 법칙은 언제 만족하는가?
1. random mating
: 위에서 언급한 allele frequency를 알아도 genotype frequency를 반드시 알 수 없다. 하지만 random mating이라면 genotype frequency의기댓값은 하디 바인 베르크에서의 값과 같다.
2. no mutation, selection, migration
: 모집단의 임의적 변화가 없어야한다.
3. 무한한 모집단 사이즈
: genetic drift가 없어야 한다.
하디 바인베르크는 위 가정들을 만족할 수 없다. 그렇다면 하디 바인베르크 법칙은 왜 필요할까? 하디 바인베르크 법칙이 중요한 이유는 그것이 귀무가설(Null hypothesis)이 될 수 있기 때문이다. 하디 바인베르크 법칙을 귀무가설로 놓고 진화적 과정이 어떻게 일어나는지를 살펴볼 수 있다.
하디 바인베르크 법칙이 깨지는 상황
하디 바인베르크 법칙은 어떠한 집단 내에서 random mating이 일어나지 않을 때이다. 즉, 인종이 섞인 집단을 하나의 집단으로 놓았을 때, 하디 바인베르크 법칙이 깨지게 된다. 예를 들어, 한국인, 중국인이 각각 하디 바인베르크 법칙을 만족하더라도, 한국인, 중국인을 합쳐서 하나의 집단을 만들고 이 집단에 대해 하디 바인베르크 법칙을 만족하는지 테스트하면 하디 바인베르크 법칙을 만족하지 않는 결과가 나올 수도 있다.
wahlund effect
인종을 합쳤을때 heterozygous genotype이 하디-바인베르크 법칙으로 구한 hetero genotype의 기댓값보다 적게 나타나는 현상.
하디 바인베르크의 법칙이 중요한 이유는 GWAS를 할 때, 연구집단이 하디-바인베르크 법칙을 만족한다는 것을 가정하기 때문이다. 하디-바인베르크 법칙이 만족되지 않으면 GWAS의 결과가 부정확할 수 있다.
예를 들어, 인종1과 인종2의 SNP의 allele frequency가 다르고, 인종1의 유병률이 인종2의 유병률보다 높다고 하자. (유전적 원인이 아닌 환경적 원인에 의해 인종1의 유병률이 높다.) 그러면 인종1이 해당 마커에 많이 갖고있는 allele 근처의 gene이 질병에 영향을 준다는 잘못된 결론을 내릴 수 있다. 그러므로 이를 방지하기 위해 하디-바인베르르 법칙을 만족하는지 테스트를 해야한다. 연구집단에 대해 하디-바인베르크 법칙을 만족한다면 그 연구집단내에서 random mating이 이일어난다는 것을 알 수 있고, 그 연구집단이 하나의 인종을 이룰 수 있다는 것을 알 수 있기 때문에 다른 환경적 요인을 보정하여 bias를 방지할 수 있다.












 Figure 1: 진핵생물에서 DNA가 단백질이 되어가는 과정
Figure 1: 진핵생물에서 DNA가 단백질이 되어가는 과정