Chip-seq 데이터를 통한 binding motif의 분석
Chip-seq 을 통해 대략적인 protein binding site (혹은 histon modification)의 시퀀스를 알고, 이 시퀀스들 중에서 비슷한 시퀀스를 찾아내어 해당 protein에 specific한 시퀀스가 무엇인지를 알아내는 분석이다. 이를 수행하는 bioinformatics Tool은 매우 많으며, [Evaluating tools for transcription factor binding site prediction] 논문에서 좋은 평가를 받았던 rGADEM을 이용하여 binding motif를 찾는 분석을 해보도록 한다. rGADEM은 GADEM 방법을 r로 구현한 것이며, GADEM은 motif를 찾는데 Genetic 알고리즘과 EM 알고리즘을 결합하여 사용한다. 자세한 방법론은 본 포스팅에서 다루지 않는다. 본 포스팅은 이 튜토리얼을 참고하였다.
# rGADEM Tutorial
# reference : https://bioconductor.org/packages/devel/bioc/vignettes/rGADEM/inst/doc/rGADEM.pdf
source("https://bioconductor.org/biocLite.R")
# biocLite("rGADEM", lib="C:/Users/Documents/R/win-library/3.4")
# biocLite("BSgenome.Hsapiens.UCSC.hg19", lib="C:/Users/Documents/R/win-library/3.4")
# biocLite("DNAStringSet", lib="C:/Users/R/win-library/3.4")
# install.packages("RCurl")
library(RCurl)
library(rGADEM)
library(BSgenome.Hsapiens.UCSC.hg19)
rGADEM은 bioLite 패키지에 있으며, 위와 같이 설치할 수 있다. rGADEM, BSgenome..., DNAStringSet 3개의 bioLite 패키지를 설치한 후, 이와 의존성이 있는 RCurl 패키지까지 설치하면 라이브러리 로드가 완료된다.
데이터
 Test_100.bed
Test_100.bed
Test_100.fasta
rGADEM을 설치하면 위 예제 데이터가 패키지 안에 기본으로 들어있다. 이 예제 데이터를 통해 실습을 해보도록 한다.
chipseq 데이터로 binding motif를 분석하기까지의 pipeline은 QC, peak calling 등의 과정을 거쳐야한다. 하지만 이번 분석에서는 peak calling까지 끝난 데이터로 분석을 한다고 가정한다. peak calling이 완료되면 protein binding이 많이 일어난 부분의 시퀀스를 얻어낼 수 있다. 이 데이터는 주로 BED 혹은 FASTA 파일로 주어진다.
1. 데이터가 BED 포맷인 경우
BED <- read.table("C:\\Test_100.bed",header=FALSE,sep="\t")
BED <- data.frame(chr=as.factor(BED[,1]),start=as.numeric(BED[,2]),end=as.numeric(BED[,3]))
BED
rgBED <-IRanges(start=BED[,2],end=BED[,3])
Sequences <- RangedData(rgBED,space=BED[,1])
rgBED
Sequences
> BED
chr start end
1 chr1 145550911 145551112
2 chr1 112321064 112321265
3 chr1 120124183 120124384
4 chr1 114560780 114560981
5 chr1 8044002 8044203
6 chr1 154708057 154708258
7 chr1 21537833 21538034
8 chr1 117197889 117198090
9 chr1 22547577 22547778
10 chr1 170982215 170982416
11 chr1 203938925 203939126
12 chr1 198607893 198608094
13 chr1 244838696 244838897
14 chr1 22613354 22613555
15 chr1 36725231 36725432
50 chr1 35024860 35025061
> rgBED
IRanges object with 50 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 145550911 145551112 202
[2] 112321064 112321265 202
[3] 120124183 120124384 202
[4] 114560780 114560981 202
[5] 8044002 8044203 202
... ... ... ...
[46] 24574361 24574562 202
[47] 46110458 46110659 202
[48] 114377432 114377633 202
[49] 152657429 152657630 202
[50] 35024860 35025061 202
> Sequences
RangedData with 50 rows and 0 value columns across 1 space
space ranges |
<factor> <IRanges> |
1 chr1 [145550911, 145551112] |
2 chr1 [112321064, 112321265] |
3 chr1 [120124183, 120124384] |
4 chr1 [114560780, 114560981] |
5 chr1 [ 8044002, 8044203] |
6 chr1 [154708057, 154708258] |
7 chr1 [ 21537833, 21538034] |
8 chr1 [117197889, 117198090] |
9 chr1 [ 22547577, 22547778] |
... ... ... ...
42 chr1 [221975293, 221975494] |
43 chr1 [ 59123669, 59123870] |
44 chr1 [ 76297843, 76298044] |
45 chr1 [188010599, 188010800] |
46 chr1 [ 24574361, 24574562] |
47 chr1 [ 46110458, 46110659] |
48 chr1 [114377432, 114377633] |
49 chr1 [152657429, 152657630] |
50 chr1 [ 35024860, 35025061] |
2. GADEM 실행
gadem <- rGADEM::GADEM(Sequences,verbose=1,genome=Hsapiens)
interation을 돌며 GADEM 알고리즘이 실행된다.
총 2개의 motif 가 생성되었다.
> length(gadem@motifList)
[1] 2
이 중에서 첫 번째 motif는 아래와 같이 생겼다.
plot(gadem@motifList[[1]])

> gadem@motifList[[1]]@alignList
[[1]]
An object of class "align"
Slot "seq":
[1] "GCCCCGACCTCTTATCTCTG"
Slot "chr":
[1] "chr1"
Slot "start":
[1] 43552181
Slot "end":
[1] 43552382
Slot "strand":
[1] "+"
Slot "seqID":
[1] 38
Slot "pos":
[1] 100
Slot "pval":
[1] 2.209965e-09
Slot "fastaHeader":
[1] 38
[[2]]
An object of class "align"
Slot "seq":
[1] "ACCCCAACCTCTTATCTCTG"
Slot "chr":
[1] "chr1"
Slot "start":
[1] 43552181
Slot "end":
[1] 43552382
Slot "strand":
[1] "+"
Slot "seqID":
[1] 38
Slot "pos":
[1] 58
Slot "pval":
[1] 2.365196e-09
Slot "fastaHeader":
[1] 38
[[3]]
An object of class "align"
Slot "seq":
[1] "ACCCCAACCCCTTATTTCTG"
Slot "chr":
[1] "chr1"
Slot "start":
[1] 43552181
Slot "end":
[1] 43552382
Slot "strand":
[1] "+"
Slot "seqID":
[1] 38
Slot "pos":
[1] 121
Slot "pval":
[1] 5.472386e-09
Slot "fastaHeader":
[1] 38
GADEM 알고리즘은 이 motif를 설명할 수 있는 총 26개의 시퀀스를 찾았다. 이 중 3개의 시퀀스가
GCCCCGACCTCTTATCTCTG
ACCCCAACCTCTTATCTCTG
ACCCCAACCCCTTATTTCTG
이다. 이러한 비슷한 시퀀스를 tfbs 라고 하며, tf protein이 이 곳에 달라붙어 regulatory role을 수행하게 된다.
[[26]]
An object of class "align"
Slot "seq":
[1] "CCTTCCTTCCTTTCTTCCTT"
Slot "chr":
[1] "chr1"
Slot "start":
[1] 200254533
Slot "end":
[1] 200254734
Slot "strand":
[1] "-"
Slot "seqID":
[1] 36
Slot "pos":
[1] 193
Slot "pval":
[1] 0.0001878598
Slot "fastaHeader":
[1] 36
위 결과처럼 - strand의 결과도 볼 수 있는데, 이 경우에는 BED파일에서 reverse strand를 보면 된다. 하지만 위 결과는 자동으로 reverse strand를 보여준다. http://genome.ucsc.edu/cgi-bin/das/hg19/dna?segment=chr1:200254533,200254734를 확인하면 위 시퀀스의 reverse인 aaggaagaaaggaaggaagg 를 확인할 수 있다.
3. INPUT이 FASTA 파일인 경우
# 2. Input이 FASTA 파일일 경우
path <- system.file("extdata/Test_100.fasta", package="rGADEM")
FastaFile <- paste(path,sep="")
Sequences <- readDNAStringSet(FastaFile, "fasta")
SequencesFasta파일의 경우 위와 같이 전처리 하면 되며, 이후의 절차는 BED 파일과 동일하다.
> Sequences
A DNAStringSet instance of length 49
width seq names
[1] 202 CTACAGCTGTTCCTTGTCATCAGCCTGGGGGTGGGTAGTATTTTGATCTTA...GTCCCCATGTGACTGTAGGGTTCCTGAAACCTGGCAGGCCACTCTGCTTG FOXA1 _ 1
[2] 202 TTATTCTGATGTGGTTTTGCGGTTATACAGTAAGCAGCACTGCTTATGTGG...ACACCAGAGGCCACCAGGACCAGAATGTTTACCAATGTAGGCAGTCACTA FOXA1 _ 10
[3] 202 AAAGGAGAAACACAGCCAAATAATAAAACAATATCTTCTGTAAGTAAAGAG...CAAAGGTGCAAAGCCCTCTTTCAGATCCATCTCCACCATTTCCCTTCAGG FOXA1 _ 11
[4] 202 TGTACCCCCCCAATATTTCATGATATTTACATGTTTGCATAGTACTTTCCT...CACAGGCAGAGTCAACATTGGAACTCGGAACACTGAGCCTGGCATTCCAA FOXA1 _ 12
[5] 202 TTTAAGACTGCCACCTGAAATCAAGTCCAGTGGCTGTTCCTGTCTTCCCGT...TTATTTAACCTTTTGCCATTAGTTTACAAGAAGACGGGTTGAGCGAGTCA FOXA1 _ 13
... ... ...
[45] 202 ATTAGTTCATGCCAGGCAGGGTTTGACCAAAACCTTGGACTCACTCCTGTC...TGCTGCTGCTAAGCCAGTGAGATAGATGGTGGTTTGGAAACACCCTCATG FOXA1 _ 5
[46] 202 CCAGAGCCACCACAGCCAGGCCTCTGCGGCCAGTGTCAACAAGGGGCCAGG...TGGGGTTTTGTTTACACACCTAGGGTCACCTGAAAACACCTGCTGTTTAA FOXA1 _ 6
[47] 202 CAGATCAGAGCCTGGGAGCGGGCCATGTGCAGCTGGATGGGCAGCTGGAGG...GCCCACCCTCCTGCCTTCCCAGCCAAGGAGGGAAGGGAGCTGTGGGAGGA FOXA1 _ 7
[48] 202 GGAATGTGATTTACCCAGATAAATCATCAGCTCAAGGGACTGCTTGGAGAG...TCCAGAGCTAGACGCAGAGGAAGGGGTCAAACATGTCCACATGGAACCTG FOXA1 _ 8
[49] 202 GGCTTTTCCAAGAACAATAGTGTTCTCCTAACAAACATTCGTTCCATCAGC...GGTGTTAGGGACAGAGTCCCAGGTGGCATAAAGTCGGGTTGGTCCTTGGC FOXA1 _ 9예제 Fasta 파일의 경우 아래와 같은 motif를 찾았고, 총 74개의 sequence가 이 consensus에 부합했다. 즉, FOXA1라는 transcription factor의 binding motif (혹은 transcription factor binding site)의 consensus를 위의 Fasta 파일의 데이터를 통해 예측해볼 수 있다.
plot(gadem@motifList[[1]])

[[72]]
An object of class "align"
Slot "seq":
[1] "CTATTGACTTT"
Slot "chr":
[1] "chr"
Slot "start":
[1] 0
Slot "end":
[1] 0
Slot "strand":
[1] "-"
Slot "seqID":
[1] 4
Slot "pos":
[1] 93
Slot "pval":
[1] 0.0001517677
Slot "fastaHeader":
[1] 4
[[73]]
An object of class "align"
Slot "seq":
[1] "ATATTTACTTG"
Slot "chr":
[1] "chr"
Slot "start":
[1] 0
Slot "end":
[1] 0
Slot "strand":
[1] "+"
Slot "seqID":
[1] 42
Slot "pos":
[1] 135
Slot "pval":
[1] 0.0001719873
Slot "fastaHeader":
[1] 42
[[74]]
An object of class "align"
Slot "seq":
[1] "CTATTTGCTGG"
Slot "chr":
[1] "chr"
Slot "start":
[1] 0
Slot "end":
[1] 0
Slot "strand":
[1] "+"
Slot "seqID":
[1] 41
Slot "pos":
[1] 53
Slot "pval":
[1] 0.0001754087
Slot "fastaHeader":
[1] 41







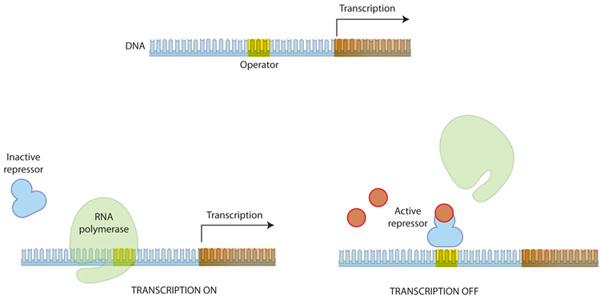
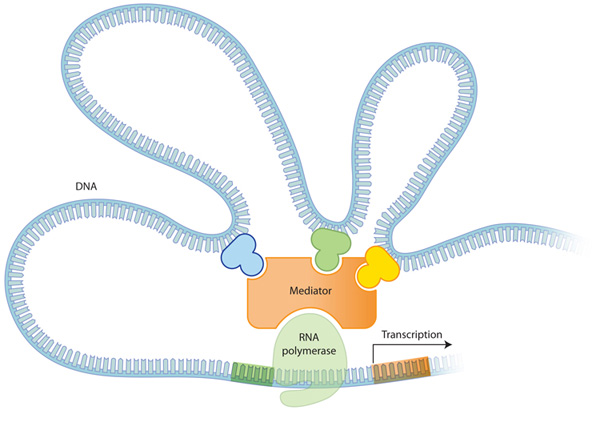
 Figure 1: 진핵생물에서 DNA가 단백질이 되어가는 과정
Figure 1: 진핵생물에서 DNA가 단백질이 되어가는 과정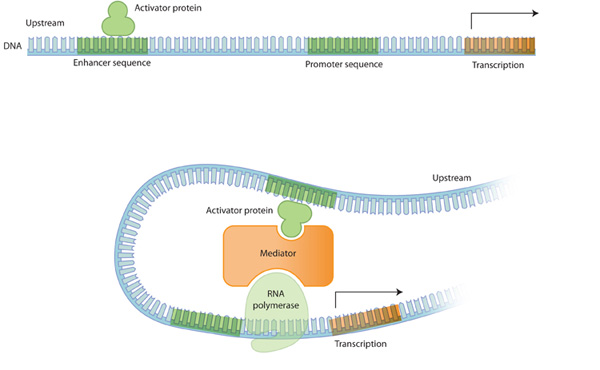



 VariantCallingLecture.pdf
VariantCallingLecture.pdf